Julia Krämer und Heinz Wiendl, Münster
Die Multiple Sklerose (MS) ist eine Erkrankung des zentralen Nervensystems (ZNS), die durch Entzündung, Demyelinisierung, Gliose und neuroaxonale Degeneration gekennzeichnet ist [47, 58, 107, 162]. Während die MS traditionell als eine hauptsächlich durch T-Zellen des adaptiven Immunsystems vermittelte Erkrankung angesehen wurde, ist mittlerweile bekannt, dass B-Zellen und fast alle Zelltypen des angeborenen Immunsystems ebenfalls eine wichtige Rolle bei der Auslösung und dem Voranschreiten der Erkrankung spielen (siehe Kasten 1 zur Erläuterung des angeborenen und adaptiven Immunsystems) [7, 10, 47, 85]. Periphere Immunzellen, die die Blut-Hirn-Schranke überwinden, verursachen Schübe und führen zur Bildung fokaler demyelinisierender Plaques [170]. Es wird angenommen, dass periphere infiltrierende Immunzellen, Endothelzellen und angeborene Immunzellen wie Mikroglia, Makrophagen und Astrozyten durch die Produktion von Zytokinen, Chemokinen, Exzitotoxinen, freien Radikalen und Komplement zur Neuroinflammation und Neurodegeneration bei der MS beitragen [50, 85, 135].
Kasten 1. Das angeborene und adaptive Immunsystem
Das Immunsystem umfasst das phylogenetisch ältere angeborene System und das adaptive System, das nur bei Wirbeltieren vorkommt [21]. Angeborene Immunzellen (z. B. Makrophagen, Mikroglia und Neutrophile) erkennen Gewebeschäden und Krankheitserreger über Mustererkennungsrezeptoren. Durch die Aktivierung dieser Rezeptoren ausgelöste Zellreaktionen sind schnell und relativ unspezifisch und umfassen Zytokinfreisetzung, Phagozytose und Zellbewegung [133, 143]. Im Gegensatz dazu steuern adaptive Immunzellen (T- und B-Zellen) spezifische und flexible Reaktionen über eine Reihe von Antigenrezeptoren und die Fähigkeit zum immunologischen Langzeitgedächtnis [21, 143].
Mikroglia bei der Multiplen Sklerose (MS)
Mikroglia sind im ZNS ansässige Makrophagen mit wichtigen Funktionen für die normale Entwicklung, Funktion und Homöostase des ZNS, einschließlich der Immunüberwachung [115]. Aktivierte Mikroglia und Makrophagen kommen in allen MS-Läsionen und in normal erscheinender weißer und grauer Substanz vor [97]. Mikroglia und Makrophagen reagieren auf viele MS-pathologische Merkmale, darunter Eisenablagerungen, meningeale Entzündungen und Fibrinogenablagerungen im ZNS infolge einer erhöhten Durchlässigkeit der Blut-Hirn-Schranke [97]. In der neuroinflammatorischen MS-Umgebung weisen aktivierte Mikroglia ein verändertes Transkriptionsprofil mit hochregulierten Entzündungs- und herunterregulierten homöostatischen Genen auf [140]. Die Reaktivität von Mikroglia und Makrophagen tritt früh bei der MS auf und ist bei progressiver MS noch ausgeprägter. Mikroglia und Makrophagen sezernieren proinflammatorische Zytokine und Chemokine, die über die Induktion der Apoptose und eine Funktionsstörung des RNA-Bindungsproteins zur Neurodegeneration beitragen können. Mikroglia und Makrophagen können auch Glutamat freisetzen und die Wiederaufnahme von Astrozyten-Glutamat hemmen, was möglicherweise zur Glutamat-Exzitotoxizität und Neurodegeneration beiträgt. Schließlich sezernieren Mikroglia und Makrophagen reaktive Sauerstoff- und Stickstoffspezies neben anderen neurotoxischen Molekülen, die eine mitochondriale Dysfunktion und oxidativen Stress auslösen [97, 195]. Mikroglia und Makrophagen haben aber auch neuroprotektive Funktionen, die Entzündungen beseitigen, Synapsen umgestalten und Schäden regenerieren können [6, 97, 123, 195]. Sie phagozytieren Trümmer von Myelin und geschädigten Zellen, sezernieren neurotrophe Faktoren, die für die Rekrutierung, das Überleben und die Reifung von Oligodendrozyten-Vorläuferzellen erforderlich sind, und unterstützen die Remyelinisierung [97, 195]. Mikroglia regulieren ihre Genexpression und ihren Phänotyp bei Bedarf dynamisch, was es ihnen ermöglicht, in unterschiedliche Funktionszustände einzutreten, um bestimmte umweltbedingte Aufgaben auszuführen [76, 195].
Während in den vergangenen 25 Jahren erhebliche Fortschritte bei der Entwicklung hochwirksamer Therapien für die schubförmige MS (RMS, relapsing multiple sclerosis) erzielt wurden, sind die Therapieoptionen für Patienten mit nicht schubförmiger progressiver MS (PMS, progressive multiple sclerosis) weiterhin begrenzt [44, 58, 111, 212]. Zu den pathologischen Merkmalen der PMS gehören eine Aktivierung von Mikroglia/Makrophagen, Astrogliose, Lymphozytenansammlungen in den Meningen und im ZNS-Parenchym, eine kortikale Demyelinisierung, mangelhafte Remyelinisierung, chronisch aktive Läsionen, diffuse neuroaxonale Schäden sowie eine Atrophie der weißen und grauen Substanz [36, 58, 111, 128, 212].
Chronisch aktive Läsionen wurden erstmals histopathologisch als Läsionen der weißen Substanz mit einem demyelinisierten hypozellulären Kern und einem Rand aus aktivierten Mikroglia und Makrophagen am Läsionsrand beschrieben [109, 110, 112]. Fortschritte in der Magnetresonanztomographie (MRT) ermöglichten die Visualisierung einer Untergruppe chronisch aktiver Läsionen in vivo, sogenannter paramagnetic rim lesions (PRLs) oder iron rim lesions (IRLs) und slowly expanding lesions (SELs) [30, 43]. PRLs und IRLs werden mit Eisen-empfindlichen MRT-Sequenzen wie der suszeptibilitätsgewichteten Bildgebung oder der T2-gewichteten Bildgebung erfasst [30] und entsprechen Eisen-beladenen aktivierten proinflammatorischen Makrophagen/Mikroglia und in viel geringerem Maße reaktiven Astrozyten [2, 43, 92].
SELs sind Bereiche, die sich graduell und konzentrisch innerhalb bestehender T2-gewichteter Läsionen ausbreiten, und durch zeitlich getrennte konventionelle MRT-Scans identifiziert werden können [55]. PRLs und SELs treten häufiger bei Patienten mit PMS auf und sind mit einem aggressiveren Krankheitsverlauf assoziiert [1, 43, 61, 124, 157], machen jedoch möglicherweise nur einen Teil aller chronisch aktiven Läsionen aus [176].
Derzeit verfügbare krankheitsmodifizierende Therapien für die MS sind in der Lage, die periphere Immunantwort zu modulieren, haben aber nur eine begrenzte Wirkung auf die im ZNS kompartimentierte Entzündung, die als treibende Kraft für die Akkumulation von Behinderungen gilt [76, 85]. Dies erklärt, warum Therapien, die Schübe und eine Schub-abhängige Verschlechterung (welche beide durch eine vorübergehende Infiltration peripherer Immunzellen bedingt sind) bei der RMS effektiv verhindern, nicht unbedingt bei der PMS wirksam sind [85]. Trotz erfolgreicher klinischer Studienergebnisse für B-Zell-depletierende Antikörpertherapien wie Rituximab, Ocrelizumab und Ofatumumab [79–81, 83, 137 ] wirken diese Wirkstoffe nur begrenzt auf das Fortschreiten der Erkrankung [68]. Weiterhin ist das Ausmaß, in dem sie in die Blut-Hirn-Schranke durchdringen und auf die kompartimentierte Entzündung bei der PMS einwirken können, begrenzt [19, 22, 105, 117]. Darüber hinaus führt die fortgesetzte Anwendung dieser nicht selektiven Therapien zu einer chronischen B-Zell-Depletion [193], was das Risiko schwerer Infektionen und möglicherweise bösartiger Erkrankungen erhöhen kann [68].
Zielstruktur Bruton-Tyrosinkinase
Um diese Einschränkungen von B-Zell-depletierenden Antikörpern zu überwinden, hat sich die jüngste Forschung auf die Hemmung der Bruton-Tyrosinkinase (BTK) konzentriert. Die BTK ist ein intrazelluläres Signalmolekül, das in B-Zellen und in den meisten hämatopoetischen Zelllinien exprimiert wird, einschließlich Monozyten, Makrophagen, Mikroglia, Mastzellen und neutrophilen Granulozyten, mit Ausnahme der natürlichen Killerzellen und vollständig differenzierten Plasmazellen [26]. Die BTK ist eine wichtige Komponente des oberen Signalwegs des B-Zell-Rezeptors (BCR) und kontrolliert die Reifung, das Überleben und die Aktivierung von B-Zellen, einschließlich der Produktion von Zytokinen und der Antigen-abhängigen Stimulation von T-Zellen [33, 166, 178, 194]. Aufgrund ihrer entscheidenden Rolle bei der B-Zell-Signalübertragung hat sich die BTK als Ziel in der Behandlung von B-Zell-Malignomen und einer wachsenden Anzahl von Autoimmunerkrankungen etabliert [33, 66]. Darüber hinaus spielt die BTK eine wichtige Rolle in Makrophagen, Mikroglia und Mastzellen; so steuert sie deren Aktivierung, die Freisetzung von Histamin durch Degranulation (bei Mastzellen), die Sekretion proinflammatorischer Zytokine sowie die Aktivierung des Inflammasoms [33, 56, 78, 91]. Unter Zellen neuronalen Ursprungs wird die BTK in Neuronen [209] und Astrozyten [132, 213] exprimiert, wohingegen in Oligodendrozyten keine BTK-Expression festgestellt wurde [132].
Die Entwicklung von BTK-Inhibitoren, die die Blut-Hirn-Schranke überwinden können, weckt die Hoffnung, dass eine neue Ära in der MS-Therapie bevorsteht, in der sowohl periphere Immunzellen, die zu MS-Schüben führen, als auch die ZNS-kompartimentierte Entzündung, die für die Akkumulation von Behinderung verantwortlich gemacht wird, gleichzeitig anvisiert werden können. Die folgende Arbeit hebt die Bedeutung der BTK für die Signalwege von B-Lymphozyten und anderen Immunzellen, die bei der MS als wichtig erachtet werden, hervor, gibt einen Überblick über die wachsende Anzahl an Daten, die den Einsatz von BTK-Inhibitoren bei der MS begründen, und erörtert die vielversprechenden Ergebnisse erster klinischer Studien.
Klinische Erfahrung mit BTK-Inhibitoren
Im Jahr 1952 beschrieb der Kinderarzt Ogden C. Bruton den Fall eines 8-jährigen Jungen mit „vollständigem Fehlen von Gammaglobulin bei ansonsten normalen Serumproteinen und wiederkehrender Pneumokokken-Sepsis“ [27]. Die von Bruton beschriebene vererbte Immunschwäche wurde als X-chromosomale Agammaglobulinämie (XLA) bezeichnet. Vier Jahrzehnte später wurde das für BTK kodierende Gen als Schlüsselmutationsstelle in der XLA identifiziert [178, 196, 202].
Seit der Entdeckung von BTK wurde eine große Anzahl von BTK-Inhibitoren für die Behandlung von B-Zell-Malignitäten entwickelt, deren Überleben bekanntermaßen eine BCR-vermittelte BTK-Signalübertragung erfordert [86]. LFM-A13 war der zuerst entwickelte BTK-Inhibitor [129], erwies sich jedoch später als stark hemmend auch für eine andere Nicht-Rezeptor-Tyrosinkinase, die Janus-Kinase 2 [104, 200]. In den folgenden Jahren wurde nach selektiveren BTK-Inhibitoren gesucht, was 2007 zur Synthese von Ibrutinib führte [153, 206]. Bisher ist Ibrutinib für die Behandlung von chronischer lymphatischer Leukämie/kleinzelligem lymphatischem Lymphom (CLL/SLL), Mantelzelllymphom (MCL), Waldenström-Makroglobulinämie, Marginalzonenlymphom und chronischer Graft-versus-Host-Krankheit [90] zugelassen; Acalabrutinib ist für die Behandlung der CLL/SLL und des MCL [29] und Zanubrutinib für die des MCL zugelassen [98]. Darüber hinaus werden BTK-Inhibitoren als mögliche Therapien für mehrere andere hämatologische Erkrankungen (Malignome und andere lymphoproliferative Erkrankungen) und nichthämatologische maligne Erkrankungen untersucht, darunter Melanome, Brust-, und Prostatakrebs, Adenokarzinome und Glioblastome sowie gegen SARS-CoV-2 (Severe acute respiratory syndrome Coronavirus-2), den Auslöser der Coronavirus-Krankheit 2019 (COVID-19) [57]. Darüber hinaus wurden oder werden BTK-Inhibitoren als potenzielle Behandlung für viele Autoimmunerkrankungen getestet, darunter rheumatoide Arthritis, Sjögren-Syndrom, systemischer Lupus erythematodes, Pemphigus vulgaris, chronische spontane Urtikaria, Asthma bronchiale und MS [164]. Die Entwicklung von BTK-Inhibitoren für die Behandlung von Autoimmunerkrankungen wurde durch die Beobachtung vorangetrieben, dass eine B-Zell-spezifische Überexpression von BTK bei Mäusen eine systemische Autoimmunität induziert [38, 103], sowie durch die Feststellung, dass die BTK-Expression in B-Zellen von Menschen mit rheumatoider Arthritis und Sjögren-Syndrom erhöht ist [39]. Aktuelle Daten zeigen, dass der klinische Nutzen von BTK-Inhibitoren bei Autoimmunerkrankungen unterschiedlich ist (keine Wirkung bei Sjögren-Syndrom und systemischem Lupus erythematodes; günstige Wirkung bei RMS, s. u.) [164].
Die Rolle der BTK in der Signalübertragung von Immunzellen
Die BTK gehört zur Tec-Familie der Nicht-Rezeptor-Tyrosinkinasen, die an der Signalvermittlung in B-Zellen und myeloischen Zellen beteiligt sind. Zu den myeloischen Zellen gehören Mikroglia, Makrophagen, Monozyten, Mastzellen, Basophile, Neutrophile und dendritische Zellen. Neben der BTK gehören noch weitere vier Mitglieder zur Tec-Familie: die Interleukin(IL)-2-induzierbare T-Zell-Kinase (ITK), die ruhende Lymphozytenkinase (RLK), die im hepatozellulären Karzinom exprimierte Tyrosinkinase (TEC) und die Knochenmark-exprimierte Kinase (BMX) [131]. Das Vorhandensein einer Pleckstrin-Homologie(PH)-Domäne ist das charakteristische Merkmal der Proteine der Tec-Familie. Die PH-Domäne ermöglicht es den Tec-Kinasen, Phosphoinositide wie Phosphatidylinositol-3,4,5-trisphosphat (PIP3) zu binden. Somit führen rezeptorvermittelte Erhöhungen der Phosphoinositidkonzentration zur Rekrutierung von Tec-Kinasen an die Plasmamembran, wo sie nachgeschaltete Signaltransduktionsmoleküle phosphorylieren können [49]. Daher unterstützt die PH-Domäne die Schlüsselrolle von Tec-Proteinen bei der Steuerung verschiedener nachgeschalteter Effektoren, die verschiedene Zellprozesse wie Wachstum, Differenzierung, Stoffwechsel, Überleben und Apoptose regulieren [54, 173, 179, 181, 198 ].
Die BTK ist entscheidend für die Entwicklung und Reifung von B-Zellen im Knochenmark, im sekundären Lymphgewebe und in der Peripherie.
Die entscheidende Rolle der BTK bei der B-Zell-Entwicklung im Knochenmark zeigt sich bei XLA-Patienten [13] und Mäusen mit X-chromosomaler Immunschwäche (XID) [160]. XLA-Patienten weisen einen nahezu vollständigen Mangel an zirkulierenden B-Zellen (< 2 % der gesamten Lymphozyten im peripheren Blut) und Plasmazellen auf [13]; die verursachenden Varianten sind Berichten zufolge über fünf strukturelle Domänen der BTK verteilt [199]. Bei den XID-Mäusen führt eine Missense-Mutation in der PH-Domäne der BTK zu einer B-Zell-Depletion, die aufgrund von Kompensationsmechanismen, die das Überleben von Prä-B-Zellen maximieren, weniger schwerwiegend ist als bei den XLA-Patienten [31, 160, 191].
In den frühen Stadien der B-Zell-Entwicklung im Knochenmark kontrolliert die BTK die Entwicklung von Prä-B-Zellen zu unreifen B-Zellen, indem es die durch IL-7 induzierte Proliferation großer zyklischer Prä-B-Zellen und ihren Übergang zu kleinen ruhenden Prä-B-Zellen sowie die Expression der Immunglobulin(Ig)-Leichtkette reguliert [134, 193, 194]. Es wurde gezeigt, dass BTK die negative Selektion autoreaktiver unreifer B-Zellen kontrolliert [103, 144]. Außerhalb des Knochenmarks ist die BTK an der Follikelmigration (über die Kontrolle der Integrinexpression) und -reifung sowie an der Aktivierung und terminalen Differenzierung von B-Zellen zu Gedächtnis-B-Zellen oder Plasmazellen beteiligt [32, 134, 144, 182, 193, 194].
Zusätzlich zur B-Zell-Signalübertragung spielt die BTK über den IgG-spezifischen Fc-Rezeptor (FcγR) eine wichtige Rolle bei der Signalvermittlung in Makrophagen und Mikroglia und über den IgE-spezifischen FcR (FcεR) in Mastzellen. Durch die Interaktion mit FcRs in myeloischen Zellen reguliert die BTK die Degranulation, die Freisetzung von Histamin und proinflammatorischen Zytokinen, die Produktion reaktiver Sauerstoffspezies (ROS) und die Aktivierung des Inflammasoms [66, 125, 166].
Abbildung 1 fasst die BTK-bezogene Signaltransduktion in B-Zellen und Makrophagen/Mikroglia zusammen. Eine detaillierte Beschreibung befindet sich im Online-Anhang zu diesem Beitrag.
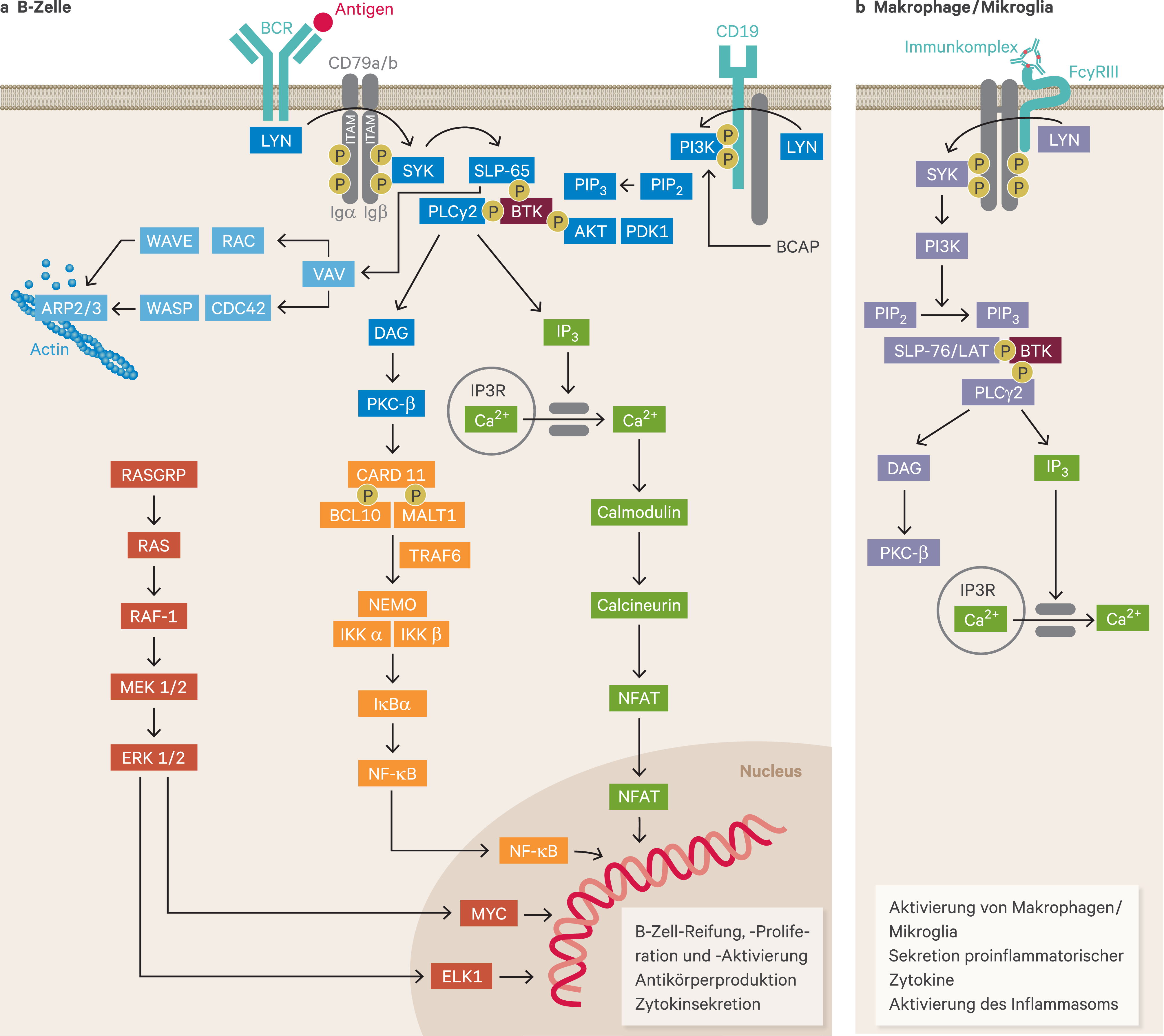
Abb. 1. Bruton-Tyrosinkinase (BTK) und Signalwege. BTK vermittelt die Signalübertragung des BCR in B-Zellen (a) und des FcγRIII in Makrophagen/Mikroglia (b) an nachgeschaltete Elemente, die für die Funktion der Immunzellen entscheidend sind. A: Die BCR-Aktivierung führt zur Bildung und Aktivierung eines Signalkomplexes, in dem BTK über das SLP-65-Gerüstprotein mit PLCγ2 verknüpft wird. Die Phosphorylierung von PLCγ2 durch BTK löst mehrere nachgeschaltete Signalwege aus, darunter die IP3R-abhängige Calciummobilisierung (grün), den Umbau des Zytoskeletts (hellblau) und die Aktivierung der NFAT- (grün), NF-κB- (orange) und RAS-Wege (rot). B: Die FcγRIII-Aktivierung rekrutiert und aktiviert LYN und SYK, die LAT phosphorylieren und einen Signalkomplex bestehend aus BTK und PLCγ2 bilden. PLCγ2 ist für die Erzeugung der sekundären Botenstoffe DAG und IP3 verantwortlich, die zelluläre Effektorreaktionen über PKC-β bzw. intrazelluläre Ca2+-Freisetzung steuern. Angepasst mit Genehmigung von Hendriks et al. [86] und Rip et al. [166]. BCAP: B-Zellen-Adapter für PI3K; BCR: B-Zell-Rezeptor; CARD11: Caspase-Rekrutierungsdomäne-enthaltendes Protein 11; DAG: Diacylglycerol; ERK: extrazelluläre signalregulierte Proteinkinase; IP3: Inositol-1,4,5-trisphosphat; IP3R: IP3-Rezeptor; ITAM: Immunrezeptor-Tyrosin-basierte Aktivierungsmotive; LAT: Linker zur Aktivierung von T-Zellen; MALT1: Mukosa-assoziiertes lymphoides Gewebe-Lymphom-Translokationsprotein 1; NFAT: Kernfaktor aktivierter T-Zellen; NF-κB: Kernfaktor Kappa B; PI3K: Phosphoinositid-3-Kinase; PIP2: Phosphatidylinositol-4,5-bisphosphat; PIP3: Phosphatidylinositol-3,4,5-trisphosphat; PKC: Proteinkinase C; PLCγ2: Phospholipase C Gamma 2; SYK: Milz-Tyrosinkinase; WASP: Wiskott-Aldrich-Syndrom-Protein
Belege für die Wichtigkeit von BTK bei der MS
Eine Reihe von aktuellen Beobachtungen an menschlichem Gewebe liefert Hinweise auf eine wichtige Rolle von BTK bei der MS, die insbesondere bei progressiver Krankheit relevant sein könnte. Das Verhältnis von phosphoryliertem BTK zu BTK-Protein, das auf eine BTK-Aktivierung hinweist, war ex vivo in Gedächtnis-B-Zellen im Blut von Patienten mit schubförmig-remittierender MS (RRMS) und sekundär progredienter MS (SPMS) im Vergleich zu gesunden Kontrollen erhöht [163]. In Gehirn-Autopsieproben von Patienten mit SPMS war die Konzentration von BTK-positiven Zellen um den Rand von Läsionen erhöht [75]*. Die Immunfärbung von Hirngewebe von Patienten mit SPMS zeigte, dass das BTK-Protein in Mikroglia exprimiert wurde, wobei eine deutliche Zunahme der Konzentration von BTK+CD68+ Zellen in Läsionen im Vergleich zu normal erscheinendem weißem Gewebe festgestellt wurde [72, 75]*. Bei der Untersuchung der Expression des BTK-Gens in MS-Läsionen wurde eine signifikante Zunahme der BTK-mRNA-Expression in Proben von primär progredienter MS (PPMS) und SPMS im Vergleich zu gesundem Gewebe festgestellt (p = 0,02). Schließlich zeigte die Einzelzell-RNA-Sequenzierung (snRNAseq) von Mikroglia aus Gehirnproben von Patienten mit PMS im Vergleich zu Kontrollgewebe eine hohe Anreicherung von BTK-mRNA als auch einer BTK-Aktivierungssignatur, die eine Hochregulation der Gene SPP1 und RGS1 und eine Herunterregulation der Gene CX3CR1 und P2RY12 umfasste [75]*.
Die veränderte Expression von CX3CR1, einem Rezeptor für den Chemokinliganden CX3CL1 [42], bei der PMS weist auf ein modifiziertes Chemokinprofil in krankheitsassoziierten Mikroglia hin. CX3CL1 hat sowohl proinflammatorische als auch antiinflammatorische Eigenschaften [42]. Ein weiteres Chemokin, CXCL13, wird stark in Mikroglia exprimiert und ist an der MS-Pathogenese beteiligt, da es bei der Rekrutierung von B-Zellen in das ZNS eine Rolle spielt [106]. Mikroglia spielen nachweislich eine Schlüsselrolle bei der Rekrutierung adaptiver Immunzellen in das ZNS [51, 127, 159]. Dieser Transport von Lymphozyten in das ZNS stellt einen plausiblen Interventionspunkt dar, der mit BTK-Inhibitoren angegangen werden könnte. Es wird angenommen, dass CXCL13 und Zytokine wie TNF-α und IL-1β, die ebenfalls von Mikroglia produziert werden [97], B-Zellen in das ZNS anziehen und halten [158]. ZNS-kompartimentierte B-Zellen wurden als einer der Haupttreiber bei der Immunpathogenese der MS identifiziert [5, 93]. Sie können das ZNS direkt schädigen durch die Sekretion von pathogenen Mikrovesikeln, die den Tod von Neuronen und Oligodendrozyten induzieren, oder durch die Freisetzung von Antikörpern, durch eine Komplementablagerung auf dem Myelin, die wiederum zu einer verstärkten Myelin-Phagozytose, der Bildung von Membranangriffskomplexen und einer B-Zell-Aktivierung führt [93].
B-Zellen können indirekt das ZNS schädigen, indem sie die Polarisierung von autoreaktiven T-Helfer-Zellen (Th) 1 und 17 innerhalb des ZNS und von der Peripherie aus vorantreiben und durch verschiedene Zytokine, die Makrophagen und Mikroglia aktivieren und die Bildung follikelähnlicher Strukturen fördern. Diese Strukturen sind mit einer ausgeprägten Demyelinisierung und Ausdünnung der Kortikalis, einer Mikroglia-Aktivierung und einer ZNS-Infiltration von T-Zellen, Makrophagen und einigen Plasmazellen verbunden [93]. Die im ZNS ansässigen B-Zellen sind vermutlich außerhalb der Reichweite von Anti-CD20-B-Zell-depletierenden Therapien wie Rituximab, Ocrelizumab und Ofatumumab. Das Potenzial von BTK-Inhibitoren, diese kompartimentierten B-Zellen zu modulieren, könnte auch bei MS einen klaren Nutzen bringen.
Präklinische Erfahrung mit BTK-Inhibitoren bei der MS
In-vitro- und In-vivo-Studien haben das Potenzial von BTK-Inhibitoren in der MS-Therapie aufgezeigt und Einblicke in mögliche Wirkmechanismen geliefert (Abb. 2). Die meisten bisherigen Erkenntnisse stammen aus Studien zu Evobrutinib und Tolebrutinib, die beide kovalente BTK-Inhibitoren sind [28, 151]. Es ist jedoch wichtig zu beachten, dass viele der Daten nur als Abstracts auf Tagungen präsentiert wurden und daher mit Vorsicht interpretiert werden sollten.

Abb. 2. Mutmaßlicher Wirkungsmechanismus von Bruton-Tyrosinkinase-(BTK-)Inhibitoren bei der Multiplen Sklerose. BTK-Inhibitoren modulieren direkt die Funktion von B-Zellen und myeloischen Zellen (einschließlich Makrophagen und Mikroglia) und zielen daher sowohl auf adaptive als auch auf angeborene immunpathogene Mechanismen der MS auf beiden Seiten der Blut-Hirn-Schranke ab [193]. Obwohl BTK-Inhibitoren die T-Zell-Funktion nicht direkt modulieren, wurde gezeigt, dass sie schädliche B-Zell-T-Zell-Wechselwirkungen behindern (nicht dargestellt) [116].
B-Zellen
B-Zellen spielen eine Schlüsselrolle bei der MS-Pathogenese und sind sowohl an Schüben als auch an der Krankheitsprogression beteiligt [34, 37, 93, 117]. BTK-Inhibitoren beeinflussen die Funktion von B-Zellen, erhalten jedoch im Gegensatz zu B-Zell-depletierenden Antikörpertherapien deren Lebensfähigkeit und Überleben [146, 194, 212]. In einer klinischen Phase-I-Studie führte die Verabreichung von BTK-Inhibitoren an gesunde Personen zu einer anfänglichen Lymphozytose, die wahrscheinlich auf eine veränderte Expression von Integrinen zurückzuführen ist [9]. Solche Veränderungen sind ein Klasseneffekt von Arzneimitteln, die auf die Signalprozesse am B-Zell-Antigenrezeptor (BCR) abzielen, und werden im Laufe der Zeit durch eine verminderte Reifung von B-Lymphozyten ausgeglichen, wobei die Zahl von CD19+-Zellen bei chronischer Exposition verringert wird [46, 88, 182]. Der Mechanismus, durch den die Behandlung mit Ibrutinib eine vorübergehende Lymphozytose und eine anhaltende Reduktion der Lymphadenopathie hervorruft [88], wurde in Primärkulturen maligner Zellen von Patienten mit CLL untersucht [46]. Diese Studie zeigte, dass die BTK-Inhibitor-Behandlung pathologisch erhöhte BCR- und Chemokin-Signalprozesse auflöste, die die Integrin-vermittelte Retention und Ansiedlung von malignen B-Zellen in Lymphknoten und Knochenmark steuern [46].
Weitere Einblicke in die funktionellen Auswirkungen der BTK-Hemmung wurden aus der pharmakodynamischen Charakterisierung des BTK-Inhibitor BIIB091 in In-vitro- und In-vivo-Studien (präklinisch und klinische Phase I) gewonnen [9]. Diese Studien zeigten, dass eine Behandlung mit BTK-Inhibitoren frühe Signalereignisse und nachgeschaltete zelluläre Effektorfunktionen in B-Zellen und myeloischen Zellen unterdrückte, einschließlich der BCR-induzierten PLC-γ2-Phosphorylierung und Antigenpräsentation für T-Zellen, Aktivierung, Proliferation und Differenzierung von B-Zellen und FcR-ausgelösten ROS-Erzeugung durch Neutrophile, TNF-α-Sekretion durch Monozyten und Basophilen-Degranulation [9]. Eine umfassende Unterdrückung BTK-abhängiger entzündlicher Aktivitäten in mehreren Immunzelltypen, einschließlich B-Zellen, Basophilen, Neutrophilen, Monozyten und Mikroglia, wurde auch in einer frühen In-vitro Studie mit Tolebrutinib berichtet [60]#.
Studien an Mausmodellen der experimentellen Autoimmunenzephalomyelitis (EAE) deuten darauf hin, dass B-Zellen ein wesentliches therapeutisches Ziel von BTK-Inhibitoren bei der MS darstellen. Evobrutinib und Tolebrutinib konnten sowohl in B-Zell- als auch in T-Zell-abhängigen EAE-Modellen dosisabhängig die Schwere der Erkrankung verbessern [14, 23, 60, 194]#. In dem B-Zell-abhängigen Modell kam es zu einer BTK-Inhibitor-induzierten Verringerung der Krankheitsschwere und zu deutlichen Veränderungen der B-Zell-Funktion, einschließlich einer Unterdrückung der Antigen-gesteuerten Aktivierung und Reifung von B-Zellen, einer BCR-induzierten proinflammatorischen Zytokinsekretion, einer Infiltration von B-Zellen in das ZNS und B-Zell-abhängige Antigenpräsentation für die Entwicklung enzephalitogener T-Zellen [194].
Der Mechanismus, der von BTK-Inhibitoren induzierten Verbesserungen im T-Zell-abhängigen EAE-Modell zugrunde liegt, ist weniger sicher, obwohl verfügbare Beweise darauf hindeuten, dass B-Zellen beteiligt sind. Die Verabreichung von Evobrutinib im T-Zell-abhängigen EAE-Modell reduzierte die leptomeningeale Entzündung, wie nach Kontrastmittelgabe im Ultrahochfeld-MRT sichtbar; histologische und immunhistochemische Untersuchungen der entzündeten Leptomeningen zeigten jedoch, dass die Anteile der B-Zellen konsistenter reduziert waren als die der T-Zellen, während myeloische Zellen persistierten [18]. Dieser Befund steht im Einklang mit primären Zellkulturexperimenten, die zeigten, dass BTK-Inhibitoren antigenspezifische proliferative und proinflammatorische Reaktionen von T-Zellen nur in Gegenwart von B-Zellen unterdrücken; dies zeigt, dass BTK-Inhibitoren auf B-Zell-T-Zell-Interaktionen abzielen [116].
Makrophagen
Mikroglia und Makrophagen spielen eine Schlüsselrolle bei der MS-Pathogenese, insbesondere im Zusammenhang mit der Förderung der Immuninfiltration und Demyelinisierung sowie der Förderung der Krankheitsprogression (siehe Kasten 1) [85, 203]. Eine Studie an XID-Mäusen zeigte eine verringerte entzündungsfördernde Funktion von Makrophagen in vitro, verbunden mit einer erhöhten Neigung zur Apoptose bei diesen Mäusen. Der potenzielle Effekt der BTK-Depletion auf Makrophagen wurde in vivo in XID-Mäusen, in denen eine Th1-abhängige EAE induziert wurde, weiter bestätigt. Diese zeigten im Vergleich zu den Wildtyp-Mäusen sowohl einen langsameren Ausbruch als auch eine geringere Schwere der Erkrankung [130]. Um mögliche nicht B-Zell-bezogene Mechanismen von Evobrutinib weiter zu untersuchen, wurden die Wirkungen dieses Wirkstoffs in einer In-vitro-Studie an Monozyten und Makrophagen untersucht. Es wurde festgestellt, dass Evobrutinib die Apoptose entzündungsfördernder Makrophagen fördert, die mit dem Granulozyten-Makrophagen-Kolonie-stimulierenden Faktor (GM-CSF) differenziert wurden. Darüber hinaus neigten Makrophagen bei gleichzeitiger Behandlung mit dem GM-CSF und Evobrutinib zu einem entzündungshemmenden Phänotyp, dessen apoptotische Zellaufnahme verstärkt war [3]#.
Mikroglia
Ergebnisse aus In-vivo-Mausstudien unterstützen den Effekt von BTK-Inhibitoren auf Mikroglia. Bei BtkE41K-Knock-in-Mäusen, die eine konstitutiv aktive Form von BTK exprimieren, konnte die Behandlung mit einem niedermolekularen BTK-Inhibitor die Verstärkung der Mikroglia-Proliferation hemmen [154]. Doppelt transgene Mäuse (Mäuse, die ein floxierbares BTK-Allel tragen, gekreuzt mit Mäusen, die die Tamoxifen-induzierbare Cre-Rekombinase unter dem Cx3CR1-Promotor tragen) entwickelten in einer Studie eine weniger schwere EAE, was auf eine Rolle von BTK in geweberesidenten myeloischen Zellen, einschließlich Mikroglia, hinweist [14]#.
Fenebrutinib 5 mg/kg zweimal täglich (BID), in vivo zwei Tage vor der EAE-Induktion (durch Immunisierung mit Peptid-Myelin-Oligodendrozyten-Glykoprotein [MOG35–55]) verabreicht, senkte nachweislich die klinische Behinderung. Dies war mit einer verringerten Mikroglia-Aktivierung verbunden, wie durch immunhistochemische Färbung des myeloischen Markers IBA1 (Ionized calcium-binding adaptor molecule 1)im Rückenmark festgestellt wurde [204]#. Im Maus-Cuprizon-Modell der Demyelinisierung (Labordiät mit 0,2 % Cuprizon über fünf Wochen) ergab die Immunhistochemie, dass BTK mit IBA1 im Hirngewebe kolokalisierte. Die Behandlung dieser Mäuse mit einem Tolebrutinib-ähnlichen BTK-Inhibitor (15 mg/kg) ergab eine BTK-abhängige Transkriptionssignatur, wobei die Interferon-Signalübertragung zu den am stärksten veränderten Signalwegen gehörte [74]#. In einer EAE-Studie (MOG35–55-Peptid-Induktion) unter Verwendung desselben Tolebrutinib-ähnlichen BTK-Inhibitors (15 mg/kg) wurden mehrere Gene, die mit krankheitsassoziierten Mikroglia assoziiert sind, nach der EAE-Induktion hochreguliert, gefolgt von einer verringerten Expression und einem reduzierten Behinderungsscore nach Behandlung mit dem BTK-Inhibitor [73]#. Darüber hinaus kehrte in vivo in einem passiven EAE-Modell, das mit enzephalitogenen T-Zellen induziert wurde, die Vorbehandlung mit Evobrutinib 10 mg/kg die Hochregulation krankheitsassoziierter Moleküle, die an der Aktivierung und Antigenpräsentation durch Mikroglia beteiligt sind, um [67]#.
Eine verminderte Mikroglia-Aktivierung wurde auch in einer In-vitro Studie beobachtet, wenn primäre Maus-Mikroglia, die einer Zytokin- und TLR-Stimulation unterzogen wurden, mit Evobrutinib [67] behandelt wurden. In einer anderen In-vitro-Studie wurde eine BTK-abhängige Transkriptionssignatur in IgG-stimulierten primären Maus-Mikroglia entdeckt [75], wobei festgestellt wurde, dass ein Tolebrutinib-ähnlicher BTK-Inhibitor die Mikroglia-Genexpression verändert und mit der Herunterregulierung proinflammatorischer Gene verbunden ist [72]#.
Ein transkriptomischer Ansatz wurde verwendet, um eine BTK-abhängige mRNA-Expressionssignatur in Mikroglia, die aus menschlichen induzierten pluripotenten Stammzellen (iPSC) gewonnen wurden, und in einer Trikultur bestehend aus menschlichen Neuronen, Astrozyten und Mikroglia zu identifizieren. Es wurde gezeigt, dass Tolebrutinib in vitro die Expression von Genen moduliert, die für proinflammatorische Zytokine und Chemokine in iPSC-abgeleiteten Mikroglia kodieren. Nachdem die proinflammatorische Signalübertragung im iPSC durch FcγR-Stimulation aktiviert wurde, modulierte die Behandlung mit Tolebrutinib diese Entzündungsreaktion, wobei eine dosisabhängige Hemmung der FcR-gesteuerten TNF-α-Sekretion berichtet wurde [72]. Entzündungssignale wurden auch in der komplexen In-vitro-Trikultur moduliert [72]. Zusammengenommen deuten diese RNAseq-Daten darauf hin, dass die Modulation von BTK in diesen Zellen mehrere für MS relevante neuroinflammatorische Wege beeinflussen kann [72]. Es ist jedoch zu berücksichtigen, dass die Ergebnisse noch nicht als peer-reviewte Veröffentlichung vorliegen (Stand 09/2023). Gemeinsamkeiten und Unterschiede zwischen den Mono- und Trikultursystemen werden derzeit untersucht.
Myelin schützen und wiederherstellen
Zusätzlich zu den Auswirkungen auf das Immunsystem gibt es zunehmend Hinweise darauf, dass die Behandlung mit BTK-Inhibitoren potenziell Vorteile für den Gewebeschutz und die Gewebereparatur hat. In einem Mausmodell der kortikalen Demyelinisierung, das mit rekombinanten Antikörpern von MS-Patienten und menschlichem Komplement induziert wurde, hemmte die Vorbehandlung mit der Tolebrutinib-ähnlichen Verbindung PRN2675 den Abbau von Myelin und die Migration von Mikroglia an die Stelle der Demyelinisierung und verhinderte den Verlust von Myelin und Oligodendrozyten [11]#. Von einem anderen BTK-Inhibitor, der Evobrutinib-ähnlichen Verbindung BTKi-1, wurde berichtet, dass er die Myelinreparatur ex vivo in organotypischen Schnittkulturen des Kleinhirns der Maus und in vivo in transgenen MBP-GFP-NTR Xenopus-Kaulquappen fördert, zwei komplementären experimentellen Modellen der De- und Remyelinisierung, die keine Zellen des adaptiven Immunsystems haben [132]. Wichtig ist, dass Evobrutinib diese Effekte in Systemen erzielte, in denen die Blut-Hirn-Schranke nicht vollständig intakt ist. Daher bleibt unklar, ob die in diesen Modellen verwendeten Konzentrationen zwischen 50 nM und 1 μM [132] im Gehirn von MS-Patienten erreicht werden können.
Klinische Erfahrung mit BTK-Inhibitoren bei der MS
Pharmakokinetik und Pharmakodynamik
Derzeit werden fünf BTK-Inhibitoren (Abb. 3) in klinischen Studien auf Sicherheit und Wirksamkeit bei Patienten mit RMS und PMS untersucht: Evobrutinib, Fenebrutinib, Remibrutinib und Tolebrutinib in Phase-III-Studien sowie Orelabrutinib in einer Phase-II-Studie (Tab. 1). Tabelle 2 und Tabelle 3 fassen die pharmakokinetischen und pharmakodynamischen Daten zu diesen Wirkstoffen zusammen, einschließlich wichtiger Aspekte wie Selektivität, Bindungsart, Zielbelegung und Grad der ZNS-Penetranz.

Abb. 3. Bruton-Tyrosinase-(BTK-)Inhibitoren, die zurzeit in klinischen Studien zur Wirksamkeit und Sicherheit bei Multipler Sklerose untersucht werden
Tab. 1. Klinische Studien zu BTK-Inhibitoren
|
Arzneistoff |
Indikation |
Aktueller Status |
Studien |
|
Evobrutinib |
RRMS |
Phase III läuft |
NCT04338061, NCT04338022 |
|
Phase III beendet |
NCT04032171, NCT04032158 |
||
|
Phase II fortlaufend |
NCT02975349 |
||
|
Rheumatoide Arthritis |
Phase II abgeschlossen |
NCT03233230, NCT02784106 |
|
|
Systemischer Lupus erythematodes |
Phase II beendet |
NCT02975336 |
|
|
Phase I vollständig |
NCT02537028 |
||
|
Tolebrutinib |
MS (einschließlich RMS, SPMS, PPMS) |
Phase III fortlaufend |
NCT04458051, NCT04411641, NCT04410991, NCT04410978 |
|
Phase II läuft |
NCT04742400, NCT03996291 |
||
|
Myasthenia gravis |
Phase III läuft |
NCT05132569 |
|
|
Fenebrutinib |
MS (einschließlich RMS, PPMS) |
Phase III läuft |
NCT04586023, NCT04586010, NCT04544449 |
|
Phase II läuft |
NCT05119569 |
||
|
Systemischer Lupus erythematodes |
Phase II abgeschlossen |
NCT02908100 |
|
|
Urtikaria |
Phase II beendet |
NCT03693625 |
|
|
B-Zell-Lymphom, chronische lymphatische Leukämie |
Phase I abgeschlossen |
NCT01991184 |
|
|
Remibrutinib |
RMS |
Phase III läuft |
NCT05156281, NCT05147220 |
|
Chronische spontane Urtikaria |
Phase III läuft |
NCT05170724, NCT05048342, NCT05032157, NCT05030311 |
|
|
Orelabrutinib |
RRMS |
Phase II läuft |
NCT04711148 |
|
Immunthrombozytopenie |
Phase II geplant |
NCT05232149, NCT05020288 |
|
|
Phase I/II geplant |
NCT05124028 |
||
|
Primäres Lymphom des Zentralnervensystems |
Phase II läuft |
NCT05209620a, NCT04899427b |
|
|
Phase I/II läuft |
NCT04961515c, NCT04831658d |
||
|
Phase I/II geplant |
NCT05021770e |
||
|
Phase I |
NCT05036577f |
||
|
Follikuläres Lymphom |
Phase II läuft |
NCT04989621g |
|
|
B-Zell-Lymphom |
Phase III läuft |
NCT05097443h |
|
|
Phase II geplant |
NCT05014100i |
||
|
Phase I/II fortlaufend |
NCT04304040j, NCT04014205 |
||
|
Non-Hodgkin-Lymphom |
Phase I/II geplant |
NCT05021770e |
|
|
Mantelzelllymphom |
Phase III geplant |
NCT05051891h |
|
|
Phase II läuft |
NCT05076097k |
||
|
Systemischer Lupus erythematodes |
Phase I/II läuft |
NCT04305197 |
BTK: Bruton-Tyrosinkinase; MS: Multiple Sklerose; PMS: primär progrediente MS; RMS: schubförmige MS; SPSM: sekundär progrediente MS
a Kombinationstherapie mit Pemetrexed. b Kombinationstherapie mit Sintilimab und Tislelizumab. c Kombinationstherapie mit Sintilimab. d Kombinationstherapie mit monoklonalem PD-1-Antikörper und Fotemustin. e Kombinationstherapie mit Thiotepa. f Kombinationstherapie mit Rituximab, Methotrexat und Dexamethason. g Kombinationstherapie mit Rituximab. h Kombinationstherapie mit Rituximab und Kombinationschemotherapie. i Kombinationstherapie mit Lenalidomid. j Kombinationstherapie mit dem rekombinanten humanisierten monoklonalen Antikörper MIL62. k Kombinationstherapie mit Lenalidomid und Rituximab
Tab. 2. Pharmakodynamik von BTK-(Bruton-Tyrosinkinase-)Inhibitoren im späten Entwicklungsstadium für die MS
|
Parameter |
Arzneistoff |
||||
|
Evobrutinib |
Tolebrutinib |
Fenebrutinib |
Remibrutinib |
Orelabrutinib |
|
|
Molekulargewicht und Summenformel |
429,5 |
455,5 |
664,8 |
507,5 |
427,5 C26H25N3O3 |
|
Wirkungsmechanismus |
Irreversible kovalente Bindung |
Irreversible kovalente Bindung |
Reversible nichtkovalente Bindung |
Irreversible kovalente Bindung |
Irreversible kovalente Bindung |
|
Kinaseselektivität* |
≥ 80 % Selektivität für BTK (90 %), BMX/ETK (89–93 %), TEC (80–82 %) < 50 % Selektivität für BLK (36–42 %), EGFR (0–1 %), ERBB2/HER2 (1 %), ITK (–2 bis 13 %), JAK3 (0 %), RLK/TXK (30–36 %) |
7 von 250 Kinasen zeigten eine > 90 %ige Hemmung mit Tolebrutinib bei 1 μM |
99 % Selektivität für BTK 56 % Selektivität für BMX/ETK ≤ 50 % Selektivität für BLK (6 %), EGFR (–7 %), ERBB2/HER2 (–1 % ), ERBB4/HER4 (1 %), ITK (–1 % ), JAK3 (5 %), RLK/TXK (4 %), TEC (23 %) |
99 % Selektivität für BTK > 50 % Selektivität für JAK3 (51 %), TEC (76 %) ≤ 50 % Selektivität für BLK (0 %), BMX/ETK (47 %), EGFR (18 %), ERBB2/HER2 (20 %), ERBB4/HER4 (0 %), ITK (12 %), RLK/TXK (10 %) |
> 90 % Selektivität für BTK mit Orelabrutinib bei 1 μM |
|
Mittlere BTK-Belegung |
[HV] |
[HV] |
ND |
[HV] |
> 99 % bei einer Dosis von 50 mg oder höher (Wirkung hielt 24 Stunden nach der Dosierung an) |
|
Daten zur Hirnpenetration (Liquor-Konzentration) |
[RMS] |
[HV] |
ND |
ND |
ND |
|
BTK IC50 |
8,9 nM (8,8 ng/ml) [28] 37,9 nM (16,3 ng/ml) [41] 58 nM (24,9 ng/ml) [77] |
0,7 nM (0,3 ng/ml) |
2,3 nM (1,5 ng/ml) [57] |
1,3 nM (0,7 ng/ ml) [57] |
1,6 nM (0,7 ng/ml) |
BID: zweimal täglich; HV: Daten von gesunden Freiwilligen; ND: keine Daten; QD: einmal täglich; RMS: Daten von Patienten mit schubförmiger Multipler Sklerose
Tab. 3. Pharmakokinetik von BTK-Inhibitoren im späten Entwicklungsstadium für die MS
|
Arzneistoff |
Bewertete Dosen |
AUC [ng x h/ml] |
Cmax [ng/ml] |
tmax [h] |
t1/2 [h] |
Referenzen |
|
|
Evobrutinib |
[HV] |
[HV] |
[HV] |
[HV] |
[16, 28, 41, 57, 77, 156] |
||
|
25 mg |
126 (46,4) |
80,4 (64,9) |
0,5 (0,3–1) |
1,59 (18,2) |
|||
|
75 mg |
345 (44,6) |
252 (60,3) |
0,5 (0,3–1) |
2,29 (18,9) |
|||
|
200 mg |
1210 (34,0) |
782 (60,1) |
0,5 (0,5–1) |
3,62 (70,5) |
|||
|
Tolebrutinib |
[HV] |
[HV] |
[HV] |
[HV] |
[151, 180] |
||
|
7,5 mg OD |
3,07 (51 %) |
1,32 (31 %) |
1,0 (0,5–2,0) |
1,57 (50 %) |
|||
|
15 mg OD |
5,85 (33 %) |
2,51 (60 %) |
1,0 (1,0–1,5) |
1,68 (21 %) |
|||
|
30 mg OD |
14,9 (75 %) |
7,46 (74 %) |
1,0 (0,5–2,0) |
2,02 (34 %) |
|||
|
60 mg OD |
18,2 (55 %) |
6,69 (66 %) |
1,5 (1,0–2,0) |
2,37 (44 %) |
|||
|
90 mg OD |
56,6 (54 %) |
21,7 (60 %) |
1,5 (1,0–2,5) |
2,83 (30 %) |
|||
|
Fenebrutinib |
[HV] |
[HV] |
[HV] |
[HV] |
[41, 57, 87] |
||
|
20 mg BID |
158 (32) |
28 (44) |
1,0 (1,0–2,0) |
–6,1 (30,7) |
|||
|
60 mg BID |
831 (28) |
217 (14) |
1,0 (1,0) |
–4,9 (9,8) |
|||
|
150 mg BID |
1695 (50) |
379 (97) |
1,0 (1,0–2,0) |
6,0 (17,7) |
|||
|
250 mg BID |
2347 (42) |
463 (62) |
1,0 (1,0–4,0) |
4,9 (31,8) |
|||
|
500 mg OD |
4241 (79) |
653 (100) |
1,0 (1,0–3,0) |
10,3 (35,1) |
|||
|
Remibrutinib |
[HV] |
[HV] |
[HV] |
[HV] |
[4, 57, 65, 99] |
||
|
100 mg OD |
488 (172) |
233 (84,1) |
0,867 (0,73–1,50) |
1,41 (1,41–11,9) |
|||
|
400 mg OD |
1300 (602) |
551 (263) |
0,758 (0,70–1,50) |
8,51 (1,22–22,3) |
|||
|
600 mg OD |
1240 (3341) |
563 (229) |
0,883 (0,50–3,00) |
8,29 (4,69–17,3) |
|||
|
100 mg BID |
518 (334) |
306 (202) |
0,775 (0,50–2,00) |
2,84 (2,15–18,9) |
|||
|
200 mg BID |
963 (439) |
347 (112) |
0,992 (0,52–2,50) |
12,4 (2,26–26,3) |
|||
|
Orelabrutinib |
ND |
ND |
ND |
ND |
[HV] ~4 |
[215] |
|
BID: zweimal täglich; OD: einmal täglich; ND: nicht bestimmt; CV: Variationskoeffizient; HV: gesunde Freiwillige; SD: Standardabweichung
BTK-Inhibitoren sind kleine Moleküle, die im Vergleich zu großen Molekülen zwei entscheidende therapeutische Vorteile haben: Sie können oral eingenommen werden und gut in die Zellen eindringen [66]. Abhängig von der Art ihrer Bindung werden irreversible oder reversible Inhibitoren unterschieden (Kasten 2) [25, 214]. Die derzeit in den Phase-II- und -III-Studien zur MS untersuchten Moleküle umfassen die irreversibel wirkenden BTK-Inhibitoren Evobrutinib, Orelabrutinib, Remibrutinib und Tolebrutinib und den reversibel wirkenden BTK-Inhibitor Fenebrutinib. Die kovalenten BTK-Inhibitoren Evobrutinib und Tolebrutinib inaktivieren die BTK irreversibel, die Signalübertragung wird jedoch nach 5–7 Tagen wiederhergestellt, da das inaktivierte Enzym durch den normalen Proteinabbau und die Proteinsynthese ersetzt wird [28, 151]. Fenebrutinib hemmt die BTK für einen relativ kurzen Zeitraum, bevor die Bindung umgekehrt wird (Verweildauer in vitro bis zu 18 Stunden) [41]. Im Allgemeinen erreichen irreversible kovalente Inhibitoren eine höhere Bindungsaffinität und eine längere Wirkungsdauer als reversible Inhibitoren, was ihre Wirksamkeit erhöhen und möglicherweise ihre erforderliche Dosis und/oder Häufigkeit der Einnahme verringern könnte [8]. Irreversible Inhibitoren können Sicherheitsprobleme im Zusammenhang mit der potenziellen Immunogenität von kovalenten Medikament-Rezeptor-Komplexen und deren Abbauprodukten mit sich bringen [8]. Die relative Wirksamkeit von BTK-Inhibitoren bei der MS wird wahrscheinlich von der Pharmakodynamik und -kinetik beeinflusst. Aktuelle Daten deuten darauf hin, dass die Potenz der BTK-Inhibition zwischen den Wirkstoffen erheblich variiert (Tab. 2), wobei Evobrutinib eine höhere Konzentration benötigt, um die halbmaximale Hemmkonzentration (IC50), einen quantitativen Index der Hemmstärke, zu erreichen, als Tolebrutinib und Fenebrutinib [164]. Es ist jedoch wichtig zu bedenken, dass sich die IC50-Werte je nach den verwendeten biochemischen und zellulären Tests unterscheiden und nicht immer die Kinasepotenz des Wirkstoffes vorhersagen, wenn sie im zellulären Kontext untersucht werden [57, 214]. Als Alternative zu IC50 wurde die halbmaximale wirksame Konzentration (EC50) gemessen, die zur Unterdrückung der B-Zell- und Basophilenaktivierung in frischen Vollblutproben von gesunden Probanden erforderlich ist. Dabei wurde festgestellt, dass die Potenz von Fenebrutinib (EC50 = 15 nM) etwas höher war als die von Tolebrutinib (EC50 = 80 nM) und weitaus höher als die von Evobrutinib (EC50 = 1271 nM) [205]. Beim Vergleich der IC50- und EC50-Werte für reversible (Fenebrutinib) und irreversible (Evobrutinib und Tolebrutinib) Kinaseinhibitoren ist jedoch zu berücksichtigen, dass irreversible Inhibitoren eine nicht-gleichgewichtige Bindungskinetik aufweisen [214].
Kasten 2. Konzepte im Zusammenhang mit Bruton-Tyrosinkinase-Inhibitoren
|
Konzept |
Beschreibung |
|
Verabreichungsweg |
Oral |
|
Bindungsmodus – Reversibilität/Irreversibilität |
Irreversible Inhibitoren stellen eine kovalente Bindung mit dem Cysteinrest 481 in der ATP-Bindungsstelle der BTK her. Reversible Inhibitoren binden nichtkovalent an eine BTK-spezifische Tasche in der SH3-Domäne über schwache, reversible Mechanismen (Wasserstoffbrücken, Ionenbindungen oder hydrophobe Wechselwirkungen), was zu einer inaktiven Kinasekonformation führt [25]. Im Allgemeinen erreichen irreversible kovalente Inhibitoren eine höhere Bindungsaffinität und eine längere Wirkungsdauer als reversible Inhibitoren, was ihre Wirksamkeit erhöhen und möglicherweise ihre erforderliche Dosis und/oder Häufigkeit der Applikation senken kann [8]. Irreversible Inhibitoren können Sicherheitsprobleme im Zusammenhang mit der potenziellen Immunogenität kovalenter Arzneimittel-Rezeptor-Komplexe und ihrer Abbauprodukte mit sich bringen [8]. |
|
Wie unterscheiden sich die BTK-Inhibitoren? |
Die BTK-Inhibitoren unterscheiden sich in ihrer Selektivität, Bindungsart, Zielbelegung, Hemmwirkung und dem Grad ihrer ZNS-Penetranz. |
|
ZNS-Penetranz |
Die ZNS-Penetranz variiert je nach BTK-Inhibitor, wobei Tolebrutinib eine größere ZNS-Penetranz aufweist als Evobrutinib und Fenebrutinib |
|
Vorteile der zweiten Generation der BTK-Inhibitoren |
Eine hohe Selektivität für eine kleine Anzahl von Kinasen verbessert die Toxizitätsprofile der BTK-Inhibitoren. |
Die ZNS-Penetranz ist eine weitere wichtige Eigenschaft von BTK-Inhibitoren bei der MS (Kasten 2). Für Evobrutinib lag die zwei bis drei Stunden nach der Einnahme gemessene Konzentration im Liquor (CSF) unter dem IC50-Wert des Wirkstoffs [156]. Im Gegensatz dazu wurde für Tolebrutinib in einer doppelblinden, Placebo-kontrollierten Phase-I-Studie gezeigt, dass die CSF-Konzentration zwei Stunden nach Verabreichung einer einzelnen 120-mg-Dosis bei gesunden Probanden den zellulären IC90-Wert überstiegen [151, 180]. Es ist jedoch möglich, dass die CSF-Konzentration von Tolebrutinib zum Zeitpunkt der Messung zwei Stunden nach Gabe einer Einzeldosis noch nicht ihren Höhepunkt erreicht hatte, da dieser Wirkstoff eine maximale Plasmakonzentration von etwa einer Stunde und eine Halbwertszeit von 90 bis 120 Minuten aufweist. Daher sind für eine umfassende Charakterisierung der CSF-Pharmakokinetik von Tolebrutinib zusätzliche Messungen zu späteren Zeitpunkten erforderlich [151].
Erste Hinweise deuten darauf hin, dass BTK-Inhibitoren eine unterschiedliche ZNS-Penetranz aufweisen, wobei Tolebrutinib im Vergleich zu Evobrutinib und Fenebrutinib eine höhere ZNS-Penetranz und Wirksamkeit und schnellere Raten der BTK-Hemmung aufweist [197]. Es ist jedoch wichtig zu bedenken, dass die gezeigten Ergebnisse bisher nur auf Konferenzen vorgestellt wurden und noch keine vollständige peer-reviewte Veröffentlichung vorliegt (Stand 09/2023).
Sicherheit und Verträglichkeit
Das Toxizitätsprofil von BTK-Inhibitoren wird sowohl von den Kinasen bestimmt, an die sie neben BTK binden (Off-Target-Effekte), als auch von On-Target-Effekten, die sich aus der Modulation des Immunsystems ergeben (z. B. ein höheres Risiko für Pilzinfektionen bei CLL-Patienten, die Ibrutinib und Acalabrutinib erhielten) [59, 120]. Als BTK-Inhibitoren der zweiten Generation weisen die derzeit für die MS-Therapie untersuchten Wirkstoffe eine hohe Selektivität für eine kleine Anzahl von Kinasen auf (Tab. 2). Dagegen binden die BTK-Inhibitoren der ersten Generation wie Ibrutinib auch an andere Kinasen der TEC-Familie und andere Tyrosinkinasen. Es wird daher angenommen, dass BTK-Inhibitoren der zweiten im Vergleich zu denen der ersten Generation weniger Off-Target-Effekte und damit ein geringeres Risiko für kardiale Toxizität oder häufig auftretende unerwünschte Ereignisse (UE) wie Herzrhythmusstörungen, Bluthochdruck, Blutungen, Durchfall, Arthralgien und Hautausschlag haben, die zum Abbruch der Behandlung führen können [25, 57, 164, 167, 187]. Das offensichtlich bessere Sicherheitsprofil der BTK-Inhibitoren der zweiten Generation wird auf eine geringere Off-Target-Hemmung von Kinasen wie der Januskinase 3, dem epidermalen Wachstumsfaktorrezeptor und potenziell anderen Mitgliedern der TEC-Familie zurückgeführt [164, 167]. Die Selektivität von Tolebrutinib für die BTK ist geringer als die von Fenebrutinib und Orelabrutinib [57].
Für Evobrutinib und Tolebrutinib wurden Daten zur Sicherheit und Wirksamkeit der Phase II veröffentlicht [138, 161]. In einer doppelblinden Phase-II-Studie zu Evobrutinib wurden 267 erwachsene Patienten mit aktiver RRMS oder SPMS mit auflagerten Schüben randomisiert, um Evobrutinib 25 mg einmal täglich (QD), 75 mg einmal oder zweimal täglich oder Placebo oder unverblindet Dimethylfumarat (DMF) 240 mg zweimal täglich für 24 Wochen zu erhalten [138]. Nach 25 Wochen wurden Patienten, die Placebo erhalten haben, für weitere 24 Wochen auf Evobrutinib 25 mg einmal täglich umgestellt; diejenigen, die bereits Evobrutinib oder DMF erhielten, setzten die ursprüngliche Behandlung fort. Nach 52 Wochen war die Behandlung mit Evobrutinib 75 mg einmal täglich (66 %) und 75 mg zweimal täglich (63 %) mit höheren Raten an unerwünschten Ereignissen assoziiert als diejenige mit Evobrutinib 25 mg (54 %) oder Placebo (56 %). Die Häufigkeit unerwünschter Ereignisse vom Grad 3–4 war unter Evobrutinib 25 mg (2 %) am niedrigsten und war in allen anderen Gruppen ähnlich hoch (11–15 %). Die höchste Rate schwerwiegender unerwünschter Ereignisse (7 %) trat bei Patienten auf, die mit Evobrutinib 75 mg zweimal täglich behandelt wurden (im Vergleich zu 4 % in allen anderen Gruppen) [138]. Die häufigsten mit der Behandlung mit Evobrutinib verbundenen unerwünschten Ereignisse waren Nasopharyngitis, erhöhte Leber- (Alanin- und Aspartat-Aminotransferase [ALT und AST]) sowie Lipasewerte [138]. Fünf (9 %) Patienten in der Placebo-Gruppe, 16 (6–13 %) in den Evobrutinib-Gruppen und zwei (4 %) in der DMF-Gruppe brachen die Studie aufgrund eines unerwünschten Ereignisses ab. Die meisten Abbrüche in den Evobrutinib-Gruppen erfolgten aufgrund erhöhter Leberwerte (alle asymptomatisch und reversibel) oder hepatobiliärer Störungen [138]. Patienten, die die doppelblinde Phase (DBP) der Phase-IIb-Studie abgeschlossen hatten, konnten an der offenen Verlängerungsphase (OLE) ab Woche 48 teilnehmen, in der sie zunächst 75 mg Evobrutinib einmal täglich erhielten, bevor sie nach einer durchschnittlichen Zeit von 49,8 ± 6,17 Wochen auf 75 mg Evobrutinib zweimal täglich umgestellt wurden [201]#. Achtzig Prozent der DBP-Patienten wurden in die OLE aufgenommen, von denen 77 % eine OLE-Behandlung von ≥ 132 Wochen abschlossen. Die Gesamtkonzentration der CD19+ B-Zellen sank während der OLE in allen Gruppen, die ursprünglich auf Evobrutinib randomisiert worden waren (0,122 × 106 Zellen/ml in der Woche 96 der OLE im Vergleich zu 0,218 × 106 Zellen/ml zu OLE-Baseline) [139]#.
Die Sicherheit von Tolebrutinib wurde in einer doppelblinden Cross-over-Dosisfindungsstudie der Phase IIb an 130 Erwachsenen mit RRMS oder schubförmiger SPMS untersucht [161]. Die Patienten wurden randomisiert, um zwölf Wochen lang täglich 5, 15, 30 oder 60 mg Tolebrutinib zu erhalten. Diese Behandlungsphase wurde von einer vierwöchigen Placebo-Phase vor oder nach der Behandlung begleitet. Über alle Dosisgruppen hinweg traten bei 54 % der Patienten Nebenwirkungen auf, deren Auftreten nicht dosisabhängig war. Die häufigsten Nebenwirkungen während der Behandlung mit Tolebrutinib waren Kopfschmerzen (insgesamt 7 %), Infektionen der oberen Atemwege (5 %) und Nasopharyngitis (4 %). Drei Patienten hatten erhöhte ALT-Spiegel, darunter zwei (in der 30-mg- und 60-mg-Gruppe) mit ALT-Konzentrationen, die mehr als das Dreifache der oberen Normgrenze betrugen. Bei einem Patienten, der 60 mg Tolebrutinib erhielt, wurde ein schwerwiegendes UE (ein schwerer MS-Schub) gemeldet. Die Behandlung wurde nicht unterbrochen, der Patient erholte sich und schloss die Studie ab. Kein Patient brach die Behandlung oder die Studie aufgrund eines UE ab [161]. Patienten, die die DBP der Phase-IIb-Studie abgeschlossen hatten, konnten am Teil A einer Langzeit-Sicherheitsstudie (LTS) teilnehmen, in der sie die Tolebrutinib-Dosis (5, 15, 30 oder 60 mg/Tag) aus der Kernstudie fortsetzten (doppelblind), und anschließend am Teil B der offenen LTS-Studie teilnahmen, in der sie 60 mg Tolebrutinib pro Tag erhielten. Nach 18 Monaten der LTS blieben 94 % der Patienten in der Studie, und die Sicherheitsdaten zeigten weiterhin eine günstige Verträglichkeit ohne das Auftreten neuer Sicherheitswarnsignale (Ergebnisse bisher nur als Konferenzpräsentation oder Abstract verfügbar) [147].
Wirksamkeit
In der 24-wöchigen randomisierten, doppelblinden Phase-II-Studie mit Evobrutinib hatten nur Patienten, die 75 mg Evobrutinib einmal täglich erhielten, in den Wochen 12 bis 24 signifikant weniger Kontrastmittel-anreichernde (KM-) Läsionen als die, die Placebo erhielten (p < 0,01) (Tab. 4). Die Baseline- und Placebo-korrigierten Raten für die Gesamtzahl der Läsionen im Zeitverlauf betrugen 1,45 (95%-KI 0,72–2,91; p = 0,32) unter Evobrutinib 25 mg, 0,30 (0,14–0,63; p = 0,005) unter Evobrutinib 75 mg einmal täglich (QD) und 0,44 (0,21–0,93; p = 0,06) unter Evobrutinib 75 mg zweimal täglich (BID) [138]. Die Patienten in der Evobrutinib-Gruppe unterschieden sich hinsichtlich der jährlichen Schubrate oder der Behinderungsprogression nicht signifikant von den Patienten in der Placebo-Gruppe. Die meisten Patienten in jeder Behandlungsgruppe waren bis zur Woche 24 schubfrei (Placebo 77 %; Evobrutinib 25 mg 74 %; Evobrutinib 75 mg QD 88 %; Evobrutinib 75 mg BID 87 %; DMF 89 %) und zeigten keine Veränderung ihrer medianen Werte auf der Expanded Disability Status Scale (EDSS) in den Wochen 24 und 48. Die Umstellung von Evobrutinib 75 mg QD auf 75 mg BID in der OLE reduzierte die jährliche Schubrate unabhängig von der Behandlung während der DBP. Die EDSS-Werte und die durchschnittliche Anzahl der KM-anreichernden Läsionen blieb in der gesamten Patientenpopulation von Woche 0 bis Woche 192 niedrig [201]#. Darüber hinaus reduzierte Evobrutinib dosisabhängig die Spiegel des Neurofilament-Leichtketten-Proteins während der DBP bis Woche 144 [108]#.
Tab. 4. BTK-Inhibitoren im Spätstadium der Entwicklung zur Behandlung der MS
|
Arzneistoff Referenz |
Verlaufstyp der MS |
Behandlungsarme |
Behandlungsdauer |
Wirksamkeitsdaten |
|
|
Klinischer Effekt |
Radiologischer Effekt |
||||
|
Evobrutinib |
|||||
|
Montalban 2019 [138] |
RMS (n = 261) |
Evobrutinib (n = 154) Dimethylfumarat (n = 54) Placebo (n = 53) |
24–48 Wochen |
↓Schübe bei EVO 75 mg einmal täglich oder zweimal täglich ↑Anteil schubfreier Patienten Keine Änderung des EDSS-Scores |
73 % ↓aller neuen KM-anreichernden Läsionen für EVO 75 mg einmal täglich (Woche 12–24); 59 % ↓aller neuen KM-anreichernden Läsionen für EVO 75 mg zweimal täglich (Woche 12–24) |
|
NCT04338061 |
RMS (n = 898) |
Evobrutinib Teriflunomid |
≤ 156 Wochen |
– |
– |
|
NCT04338022 |
RMS (n = 898) |
Evobrutinib Teriflunomid |
≤ 156 Wochen |
||
|
Tolebrutinib |
|||||
|
Reich 2021 [161] |
RMS (n = 130) |
Tolebrutinib |
12 Wochen |
Nicht berichtet |
85 % ↓neue KM-anreichernde Läsionen |
|
NCT04458051 |
PPMS (n = 990) |
Tolebrutinib Placebo |
24–48 Monate |
– |
– |
|
NCT04411641 |
nrSPMS (n = 1290) |
Tolebrutinib Placebo |
24–48 Monate |
||
|
NCT04410991 |
RMS (n = 900) |
Tolebrutinib Teriflunomid |
18–36 Monate |
||
|
NCT04410978 |
RMS (n = 900) |
Tolebrutinib Teriflunomid |
18–36 Monate |
||
|
NCT04742400 |
MS (n = 10) |
Tolebrutinib |
≥ 96 Wochen |
||
|
Fenebrutinib |
|||||
|
NCT05119569 |
RMS (n = 102) |
Fenebrutinib Placebo |
12 Wochen |
– |
– |
|
NCT04586023 |
RMS (n = 736) |
Fenebrutinib Teriflunomid |
96 Wochen |
||
|
NCT04586010 |
RMS (n = 736) |
Fenebrutinib Teriflunomid |
96 Wochen |
||
|
NCT04544449 |
PPMS (n = 946) |
Fenebrutinib Ocrelizumab |
120 Wochen |
||
|
Remibrutinib |
|||||
|
NCT05147220 |
RMS (n = 800) |
Remibrutinib Teriflunomid |
≤ 30 Monate |
– |
– |
|
NCT05156281 |
RMS (n = 800) |
Remibrutinib Teriflunomid |
≤ 30 Monate |
||
|
Orelabrutinib |
|||||
|
NCT04711148 |
RRMS (n = 160) |
Orelabrutinib Placebo |
120 Wochen |
– |
– |
KM: Kontrastmittel; EDSS: Expanded Disability Status Scale; EVO: Evobrutinib; nrSPMS: nicht-schubförmige sekundär progrediente MS; PPMS: primär progressive MS; RMS: schubförmige MS; RRMS: schubförmig-remittierende MS
In der zwölfwöchigen Phase-IIb-Studie mit Tolebrutinib wurde eine dosisabhängige Wirkung auf die Bildung KM-anreichernder Läsionen festgestellt (p = 0,03), wobei die 60-mg-Tolebrutinib-Dosis im Vergleich zu Placebo die Bildung neuer Läsionen nach zwölf Wochen Behandlung um 85 % (95%-KI 28–97) reduzierte; 90 % der Patienten in der 60-mg-Gruppe hatten keine neuen Läsionen, wohingegen 75 % der Patienten nach vier Wochen Placebo keine neuen Läsionen hatten. Es wurde auch eine dosisabhängige Wirkung auf die Anzahl neuer/sich vergrößernder T2-Läsionen festgestellt (p < 0,01), wobei die 60-mg-Dosis zu einer relativen Reduktion um 89 % (95%-KI 68–96) im Vergleich zu Placebo führte [161]. Sowohl die Wirksamkeits- als auch die Sicherheitsergebnisse der zwölfwöchigen Kernstudie waren in der Gesamtpopulation als auch in der Untergruppe der Patienten mit hochaktiver Erkrankung konsistent [186]. In der Gesamtpopulation blieb nach 18 Monaten im LTS die Anzahl der neuen KM-anreichernden Läsionen im 60-mg-Tolebrutinib-Arm niedrig. In der LTS-Woche 72 blieben 84,7 % der Patienten schubfrei und die jährliche Schubrate war niedrig. Die EDSS-Werte blieben auch in den ersten 18 Monaten des LTS stabil [147]#.
Zusammenfassung
Immer mehr Daten weisen darauf hin, dass BTK-Inhibitoren im Gegensatz zu den derzeit zugelassenen Immuntherapien das Potenzial haben, die Krankheitsprogression bei der RMS und PMS positiv zu beeinflussen [212]. Als kleine Moleküle können BTK-Inhibitoren die Blut-Hirn-Schranke überwinden und somit gleichzeitig auf Zellen des adaptiven und angeborenen Immunsystems im peripheren und zentralen Nervensystem einwirken. BTK-Inhibitoren können das entzündungfördernde pathogene Potenzial von Zellen des angeborenen Immunsystems zugunsten eines immunregulatorischen Phänotyps dämpfen und/oder den Schutz und die Reparatur der Myelinschicht fördern. Darüber hinaus haben sie gegenüber B-Zell-depletierenden monoklonalen Antikörpern den Vorteil, die Funktion myeloider Zellen modulieren zu können und im ZNS verfügbar zu sein, wie für einige Inhibitoren nachgewiesen werden konnte [33, 40, 113, 194].
Ob die Wirksamkeit von BTK-Inhibitoren jedoch die von monoklonalen Antikörpern übertrifft, muss noch ermittelt werden [40]. Ein Großteil der präklinischen Daten zur Wirkung von BTK-Inhibitoren auf Mikroglia wurde bisher nur als Abstract veröffentlicht; daher müssen diese Ergebnisse mit Vorsicht betrachtet werden. Darüber hinaus bleibt unklar, ob die in vitro verwendeten BTK-Inhibitor-Konzentrationen auch in vivo beim Menschen erreichbar sind.
Ein möglicher Nachteil von B-Zell-depletierenden Therapien besteht darin, dass Patienten möglicherweise chronisch der potenziellen Vorteile regulatorischer B-Zellen beraubt werden [82]. Ob BTK-Inhibitoren hier Abhilfe schaffen können, ist ungewiss, da festgestellt wurde, dass Ibrutinib die Produktion von immunsuppressivem Adenosin in regulatorischen B-Zellen von Krebspatienten reduziert [95]. Darüber hinaus bleibt abzuklären, wo BTK-Inhibitoren in Bezug auf die Behandlungssequenz bei der MS eine Rolle spielen. Obwohl die Reversibilität der Wirkungen von BTK-Inhibitoren vielversprechend ist, ist nicht zu erwarten, dass sie Antigen-erfahrene (einschließlich autoreaktive) Zellen des adaptiven Immunsystems vollständig auslöschen. Andererseits eröffnet dies möglicherweise eine elegante Möglichkeit, die Effekte von immundepletierenden Therapien aufrechtzuerhalten, wenn BTK-Inhibitoren während der Repopulation angewendet werden [126, 169].
Bisher haben zwei Phase-II-Studien gezeigt, dass BTK-Inhibitoren die Krankheitsaktivität bei Patienten mit RMS bei günstigen Verträglichkeits- und Sicherheitsprofilen begrenzen können [138, 161]. Auch wenn die schwerwiegenden Nebenwirkungen von BTK-Inhibitoren, die bei hämatologischen Erkrankungen berichtet wurden, bei der MS noch nicht gemeldet wurden, ermöglichen die geringe Stichprobengröße und die kurzen Beobachtungszeiträume von Phase-II-Studien keine umfassende Bewertung diese Aspekte. Das Sicherheitsprofil von BTK-Inhibitoren kann bei der MS und anderen mit diesen Wirkstoffen behandelten Krankheiten aufgrund unterschiedlicher Komorbiditäten und pathophysiologischer Prozesse variieren [183]. Derzeit laufen mehrere klinische Studien, die robustere Daten zur Langzeitwirksamkeit und -sicherheit dieser Wirkstoffe bei der MS liefern werden. Es ist zu erwarten, dass Unterschiede in den pharmakologischen Eigenschaften von BTK-Inhibitoren (Pharmakodynamik, Selektivität und ZNS-Penetration) zu unterschiedlichen Wirksamkeits- und Sicherheitsprofilen in Phase-III-Studien und in der klinischen Praxis führen können [57, 167].
In künftigen Studien sollte weiter untersucht werden, ob BTK-Inhibitoren weitere Kriterien erfüllen könnten, die bei neuen MS-Therapien wünschenswert wären, insbesondere die Reduktion von oxidativem Stress, die Bindung von Eisen und die Förderung der Remyelinisierung [212]. Es wird auch von Interesse sein, die relativen Eigenschaften der verschiedenen Arten von BTK-Inhibitoren zu bestimmen, beispielsweise ob eine seltenere Verabreichung von kovalent bindenden BTK-Inhibitoren zu einer verbesserten Arzneimitteleinnahme führen könnte [15]. Weiterhin könnte ein sequenzieller Behandlungsansatz untersucht werden, bei dem auf die anfängliche Behandlung mit einer derzeit zugelassenen Antikörpertherapien eine Therapie mit einem BTK-Inhibitor folgt [66]. Um herauszufinden, ob diese Verabreichungsreihenfolge am effektivsten ist, ist eine klinische Bewertung erforderlich. Im Bereich der Onkologie wurde festgestellt, dass die Behandlung mit BTK-Inhibitoren den Austritt von Lymphozyten aus der Milz und den Lymphknoten in das periphere Blut auslöst. Diese massive Pro-B-Zell-Lymphozytose könnte es den Anti-CD20-Therapien ermöglichen, neoplastische Zellen zu eliminieren, die sich andernfalls „okkult“ in sekundären lymphatischen Organen befinden könnten [192]. Eine weitere wichtige Überlegung ist, dass B-Zellen und Mikroglia zwar als wichtige therapeutische Ziele von BTK-Inhibitoren gelten, der tatsächliche Beitrag dieser Zellen zur MS-Pathophysiologie jedoch nicht vollständig geklärt ist; beispielsweise müssen die Prozesse, die dazu führen, dass Mikroglia bei der MS schädliche oder vorteilhafte Phänotypen annehmen, weiter untersucht werden [116, 195]. Ebenso ist die Interaktion zwischen B-Zellen und T-Zellen ein weiteres therapeutisches Ziel von BTK-Inhibitoren, das zunehmend als Teil der MS-Pathophysiologie betrachtet wird [10, 116].
Aktuelle Daten zeigen, dass Mikroglia unter physiologischen Bedingungen an der Regulierung der Gewebehomöostase beteiligt sind und im verletzten ZNS eine entzündungshemmende und potenziell regenerative Rolle spielen können (Kasten 1) [76]. Neben ihren pathogenen Eigenschaften können B-Zellen oder bestimmte B-Zell-Subpopulationen wahrscheinlich immunologisch ausgleichende Funktionen ausüben, um die Gewebeentzündungen und die entzündungsfördernde Aktivierung anderer Immunzellen einzuschränken [82]. Eine zentrale Herausforderung zukünftiger Forschung wird darin bestehen, die Reaktionsmuster von B-Zellen und anderen Immunzellen, insbesondere von Mikroglia, in verschiedenen Kontexten weiter zu entschlüsseln; beispielsweise könnte die Mikroglia-Aktivierung unter BTK-Inhibitoren in vivo mit der Positronenemissionstomographie und Radioliganden abgebildet werden [185]. Solche Erkenntnisse könnten es ermöglichen, die vorteilhaften Eigenschaften des Immunsystems zu nutzen und gleichzeitig schädliche Reaktionen zu hemmen [195].
Wichtige Punkte
Die Bruton-Tyrosinkinase (BTK) ist ein intrazelluläres Signalmolekül, das die Reifung, Proliferation, das Überleben und die Aktivierung von B-Zellen und myeloischen Zellen reguliert.
Aufgrund ihrer Fähigkeit, gleichzeitig auf adaptive und angeborene Immunmechanismen in der Peripherie und im ZNS abzuzielen, sind BTK-Inhibitoren ein vielversprechender therapeutischer Ansatz zur Behandlung der RMS und PMS.
Präklinische Studien zeigen, dass die BTK-Hemmung wichtige pathologische Merkmale der MS unterdrücken kann, darunter die B-Zell-Aktivierung, Infiltration von ZNS-Lymphozyten, leptomeningeale Entzündung, proinflammatorische Mikroglia-Aktivierung und Demyelinisierung.
Fünf BTK-Inhibitoren, die sich in der Selektivität, der Stärke der BTK-Hemmung, der ZNS-Penetranz und der Bindungsart unterscheiden, werden derzeit in klinischen Studien auf ihre Wirksamkeit und Sicherheit bei Patienten mit schubförmiger und progressiver MS untersucht.
Danksagungen
Dieser Übersichtsartikel wurde vom SFB-SFB TR128 „Initiating Effector versus Regulatory Mechanisms in Multiple Sclerosis – Progress into Tackling the Disease“ der Deutschen Forschungsgemeinschaft (DFG) gefördert (A09 an Heinz Wiendl, Z02 an Heinz Wiendl).
Wir danken Prof. Dr. Amit Bar-Or (Center for Neuroinflammation and Neurotherapeutics und Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia) und Dr. Timothy J. Turner (Sanofi, Cambridge) für ihre Unterstützung. Der vorliegende Artikel basiert auf einer englischsprachigen Übersicht, die wir gemeinsam mit ihnen veröffentlicht haben (Nat Rev Neurol 2023;19:289–304).
Interessenkonflikterklärung
JK erhielt Honorare für Vorträge von Biogen, Merck, Mylan, Novartis, Sanofi, Roche und Teva sowie finanzielle Forschungsunterstützung von Amicus Therapeutics und Sanofi.
HW erhielt Honorare für seine Tätigkeit als Mitglied wissenschaftlicher Beiräte von Abbvie, Alexion, Argenx, Bristol Myers Squibb/Celgene, Janssen, Merck, Novartis. Er erhält Referentenhonorare und Reiseunterstützung von Alexion, Biogen, Bristol Myers Squibb, F. Hoffmann-La Roche Ltd., Genzyme, Merck, Neurodiem, Novartis, Roche Pharma AG, TEVA, WebMD Global. Er fungiert als bezahlter Berater für Abbvie, Actelion, Argenx, Biogen, Bristol Myers Squibb, EMD Serono, Fondazione Cariplo, Gossamer Bio, Idorsia, Immunic, Immunovant, Janssen, Lundbeck, Merck, NexGen, Novartis, PSI CRO, Roche, Sanofi, Schweizerische Gesellschaft für Multiple Sklerose, UCB und Worldwide Clinical Trials. Seine Forschung wird durch das Bundesministerium für Bildung und Forschung (BMBF), die Deutsche Forschungsgesellschaft (DFG), die Deutsche Myasthenie Gesellschaft eV, Alexion, Amicus Therapeutics Inc., Argenx, Biogen, CSL Behring, F. Hoffmann-La Roche, Genzyme, Merck KgaA, Novartis Pharma, Roche Pharma und UCB Biopharma gefördert.
* Die Ergebnisse liegen noch nicht als peer-reviewte Veröffentlichung vor (Stand 09/2023)
# Die Ergebnisse sind bisher nur als Konferenzpräsentation oder Abstract verfügbar.
Literatur
1. Absinta M, Dal-Bianco A. Slowly expanding lesions are a marker of progressive MS – Yes. Mult Scler 2021;27:1679–81, doi: 10.1177/13524585211013748.
2. Absinta M, et al. A lymphocyte–microglia–astrocyte axis in chronic active multiple sclerosis. Nature 2021;597:709–14, doi: 10.1038/s41586-021-03892-7.
3. Alankus Y, Grenningloh R, Haselmayer P, Bender A, et al. BTK inhibition prevents inflammatory macrophage differentiation: a potential role in MS. Mult Scler 2018;24(Suppl 2):P557.
4. Angst D, et al. Discovery of LOU064 (remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s tyrosine kinase. J Med Chem 2020;63:5102–18, doi: 10.1021/acs.jmedchem.9b01916.
5. Arneth BM. Impact of B cells to the pathophysiology of multiple sclerosis. J Neuroinflammation 2019;16:128, doi: 10.1186/s12974-019-1517-1.
6. Baaklini CS, Rawji KS, Duncan GJ, Ho MFS, et al. Central nervous system remyelination: roles of glia and innate immune cells. Front Mol Neurosci 2019;12:225, doi: 10.3389/fnmol.2019.00225.
7. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018;97:742–68, doi: 10.1016/j.neuron.2018.01.021.
8. Baillie TA. Targeted covalent inhibitors for drug design. Angew Chem Int Ed Engl 2016;55:13408–21, doi: 10.1002/ange.201601091.
9. Bame E, et al. Next-generation Bruton’s tyrosine kinase inhibitor BIIB091 selectively and potently inhibits B cell and Fc receptor signaling and downstream functions in B cells and myeloid cells. Clin Transl Immunol 2021;10:e1295, doi: 10.1002/cti2.1295.
10. Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol 2021;20:470–83, doi: 10.1016/s1474-4422(21)00063-6.
11. Barr H, et al. Microglial BTK signaling regulates immune-mediated cortical demyelination. Mult Scler 2021;27(Suppl 1):P196.
12. Barrington RA, Borde M, Rao A, Carroll MC. Involvement of NFAT1 in B cell self-tolerance. J Immunol 2006;177:1510–5, doi: 10.4049/jimmunol.177.3.1510.
13. Basile N, et al. Clinical and molecular analysis of 49 patients with X-linked agammaglobulinemia from a single center in Argentina. J Clin Immunol 2009;29:123–9, doi: 10.1007/s10875-008-9227-y.
14. Bassani C, et al. The role of human and mouse BTK in myeloid cells (P3–4.006). Neurology 2022;98:2707.
15. Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today 2015;20:1061–73, doi: 10.1016/j.drudis.2015.05.005.
16. Becker A, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration-QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin Transl Sci 2020;13:325–36, doi: 10.1111/cts.12713.
17. Berland R, Wortis HH. Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proc Natl Acad Sci USA 2003;100:13459–64, doi: 10.1073/pnas.2233620100.
18. Bhargava P, et al. Imaging meningeal inflammation in CNS autoimmunity identifies a therapeutic role for BTK inhibition. Brain 2021;144:1396–408, doi: 10.1093/brain/awab045.
19. Bhargava P, et al. Trial of intrathecal rituximab in progressive multiple sclerosis patients with evidence of leptomeningeal contrast enhancement. Mult Scler Relat Disord 2019;30:136–40, doi: 10.1016/j.msard.2019.02.013.
20. Bhattacharyya S, et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin–NFAT signaling network. J Exp Med 2011;208:823–39, doi: 10.1084/jem.20100945.
21. Bonilla FA, Oettgen HC. Adaptive Immunity. J Allergy Clin Immunol 2010;125:S33–40, doi: 10.1016/j.jaci.2009.09.017.
22. Bonnan M, et al. No early effect of intrathecal rituximab in progressive multiple sclerosis (EFFRITE Clinical Trial). Mult Scler Int 2021;2021:8813498, doi: 10.1155/2021/8813498.
23. Boschert U, et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by evobrutinib (M2951): a novel Bruton’s tyrosine kinase inhibitor. Mult Scler 2017;23(Suppl 3):P678.
24. Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure 2011;19:447–59, doi: 10.1016/j.str.2011.02.004.
25. Brullo C, Villa C, Tasso B, Russo E, et al. BTK inhibitors: a medicinal chemistry and drug delivery perspective. Int J Mol Sci 2021;22:7641, doi: 10.3390/ijms22147641.
26. Brunner C, Müller B, Wirth T. Bruton’s tyrosine kinase is involved in innate and adaptive immunity. Histol Histopathol 2005;20:945–55, doi: 10.14670/hh-20.945.
27. Bruton OC. Agammaglobulinemia. Paediatrics 1952;9:722–8, doi: 10.1542/peds.9.6.722.
28. Caldwell RD, et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem 2019;62:7643–55, doi: 10.1021/acs.jmedchem.9b00794.
29. Calquence (Acalabrutinib)-Kapseln [US-Fachinformation]. (AstraZeneca, Wilmington, DE, 2019).
30. Calvi A, et al. In vivo imaging of chronic active lesions in multiple sclerosis. Mult Scler 2022;28:683–90, doi: 10.1177/1352458520958589.
31. Cancro MP, et al. xid mice reveal the interplay of homeostasis and Bruton’s tyrosine kinase-mediated selection at multiple stages of B cell development. Int Immunol 2001;13:1501–14, doi: 10.1093/intimm/13.12.1501.
32. Cariappa A, et al. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, BTK, and CD21. Immunity 2001;14:603–15, doi: 10.1016/s1074-7613(01)00135-2.
33. Carnero Contentti E, Correale J. Bruton’s tyrosine kinase inhibitors: a promising emerging treatment option for multiple sclerosis. Exp Opin Emerg Drugs 2020;25:377–81, doi: 10.1080/14728214.2020.1822817.
34. Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, et al. B cells in multiple sclerosis — from targeted depletion to immune reconstitution therapies. Nat Rev Neurol 2021;17:399–414, doi: 10.1038/s41582-021-00498-5.
35. Chiu CW, Dalton M, Ishiai M, Kurosaki T, et al. BLNK: molecular scaffolding through ‘cis’-mediated organization of signaling proteins. EMBO J 2001;21:6461–72, doi: 10.1093/emboj/cdf658.
36. Choi SR, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012;135:2925–37, doi: 10.1093/brain/aws189.
37. Comi G, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol 2021;89:13–23, doi: 10.1002/ana.25927.
38. Corneth OB, et al. Enhanced expression of Bruton’s tyrosine kinase in B cells drives systemic autoimmunity by disrupting T cell homeostasis. J Immunol 2016;197:58–67, doi: 10.4049/jimmunol.1600208.
39. Corneth, OBJ, et al. Enhanced Bruton’s tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol 2017;69:1313–24, doi: 10.1002/art.40059.
40. Correale J. BTK inhibitors as potential therapies for multiple sclerosis. Lancet Neurol 2021;20:689–91, doi: 10.1016/S1474-4422(21)00250-7.
41. Crawford JJ, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J Med Chem 2018;61:2227–45, doi: 10.1021/acs.jmedchem.7b01712.
42. Cui LY, Chu SF, Chen NH. The role of chemokines and chemokine receptors in multiple sclerosis. Int Immunopharmacol 2020;83:106314, doi: 10.1016/j.intimp.2020.106314.
43. Dal-Bianco A, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol 2017;133:25–42, doi: 10.1007/s00401-016-1636-z.
44. Dangond F, et al. Facing the urgency of therapies for progressive MS — a progressive MS alliance proposa. Nat Rev Neurol 2021;17:185–92, doi: 10.1038/s41582-020-00446-9.
45. David L, et al. Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc Natl Acad Sci USA 2018;115:1499–504, doi: 10.1073/pnas.1721967115.
46. de Rooij MF, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012;119:2590–4, doi: 10.1182/blood-2011-11-390989.
47. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol 2015;15:545–58, doi: 10.1038/nri3871.
48. Di Paolo JA, et al. Specific BTK inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol 2011;7:41–50, doi: 10.1038/nchembio.481.
49. Dickson EJ, Hille B. Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem J 2019;476:1–23, doi: 10.1042/bcj20180022.
50. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem 2016;139(Suppl 2):136–53, doi: 10.1111/jnc.13607.
51. Dong Y, Yong VW. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat Rev Neurol 2019;15:704–17, doi: 10.1038/s41582-019-0253-6.
52. Doyle SL, Jefferies CA, Feighery C, O’Neill LA. Signaling by Toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J Biol Chem 2007;282:36953–60, doi: 10.1074/jbc.M707682200.
53. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, et al. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 2002;418:790–3, doi: 10.1038/nature00859.
54. Ekman N, et al. The BMX tyrosine kinase is activated by IL-3 and G-CSF in a PI-3K dependent manner. Oncogene 2000;19:4151–8, doi: 10.1038/sj.onc.1203763.
55. Elliott C, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesion. Mult Scler 2019;25:1915–25, doi: 10.1177/1352458518814117.
56. Ellmeier W, Abramova A, Schebesta A. Tec family kinases: regulation of FcεRI-mediated mast-cell activation. FEBS J 2011;278:1990–2000, doi: 10.1111/j.1742-4658.2011.08073.x).
57. Estupinan HY, Berglof A, Zain R, Smith CIE. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front Cell Dev Biol 2021;9:630942, doi: 10.3389/fcell.2021.630942.
58. Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov 2019;18:905–22, doi: 10.1038/s41573-019-0035-2.
59. Fiorcari S, et al. BTK inhibition impairs the innate response against fungal infection in patients with chronic lymphocytic leukemia. Front Immunol 2020;11:2158, doi: 10.3389/fimmu.2020.02158.
60. Francesco M, et al. PRN2246, a potent and selective blood–brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Mult Scler 2017;23(Suppl 3):P989.
61. Frischer JM, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015;78:710–21, doi: 10.1002/ana.24497.
62. Fujimoto M, et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity 2000;13:47–57, doi: 10.1016/s1074-7613(00)00007-8.
63. Furumoto Y, Nunomura S, Terada T, Rivera J, et al. The FcεRIβ immunoreceptor tyrosine-based activation motif exerts inhibitory control on MAPK and IκB kinase phosphorylation and mast cell cytokine production. J Biol Chem 2004;279:49177–87, doi: 10.1074/jbc.M404730200.
64. Fütterer K, Wong J, Grucza RA, Chan AC, et al. Structural basis for Syk tyrosine kinase ubiquity in signal transduction pathways revealed by the crystal structure of its regulatory SH2 domains bound to a dually phosphorylated ITAM peptide. J Mol Biol 1998;281:523–37, doi: 10.1006/jmbi.1998.1964.
65. Gabizon R, London N. A fast and clean BTK inhibitor. J Med Chem 2020;63:5100–1, doi: 10.1021/acs.jmedchem.0c00597.
66. Garcia-Merino A. Bruton’s tyrosine kinase inhibitors: a new generation of promising agents for multiple sclerosis therapy. Cells 2021;10:2560, doi: 10.3390/cells10102560.
67. Geladaris A, et al. Targeting BTK in chronic CNS autoimmunity inhibits activation of microglia. Mult Scler 2021;27(Suppl 2):P971.
68. Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20(+) B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics 2017;14:835–41, doi: 10.1007/s13311-017-0557-4.
69. Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol 2010;2:a000182, doi: 10.1101/cshperspect.a000182.
70. Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol 2006;6:218–30, doi: 10.1038/nri1782.
71. Gray P, et al. MyD88 adapter-like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signal transduction. J Biol Chem 2006;281:10489–95, doi: 10.1074/jbc.M508892200.
72. Gruber R, et al. Evaluating the effect of BTK inhibitor tolebrutinib in human microglia. Mult Scler 2021;27(Suppl 2):P391.
73. Gruber R, et al. Evaluating the effect of a Bruton’s tyrosine kinase inhibitor in a murine experimental autoimmune encephalomyelitis model of multiple sclerosis. Mult Scler 2022;28(Suppl_3):P174.
74. Gruber R, et al. Decoding Bruton’s tyrosine kinase signaling in neuroinflammation. Mult Scler 2020;26(3_suppl):P0311.
75. Gruber R, et al. Central effects of BTK inhibition in neuroinflammation. Neurology 2020;94(Suppl 15):808.
76. Guerrero BL, Sicotte NL. Microglia in multiple sclerosis: friend or foe? Front Immunol 2020;11:374, doi: 10.3389/fimmu.2020.00374.
77. Haselmayer P, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease model. J Immunol 2019;202:2888–906, doi: 10.4049/jimmunol.1800583.
78. Hata D, et al. Involvement of Bruton’s tyrosine kinase in FcεRI-dependent mast cell degranulation and cytokine production. J Exp Med 1998;187:1235–47, doi: 10.1084/jem.187.8.1235.
79. Hauser SL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosi. N Engl J Med 2008;358:676–88, doi: 10.1056/NEJMoa0706383.
80. Hauser SL, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017;376:221–34, doi: 10.1056/NEJMoa1601277.
81. Hauser SL, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med 2020;383:546–57, doi: 10.1056/NEJMoa1917246.
82. Häusser-Kinzel S, Weber MS. The role of B cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorder. Front Immunol 2019;10:201, doi: 10.3389/fimmu.2019.00201.
83. Hawker K, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009;66:460–71, doi: 10.1002/ana.21867.
84. Haxhinasto SA, Bishop GA. Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J Biol Chem 2004;279:2575–82, doi: 10.1074/jbc.M310628200.
85. Healy LM, Stratton JA, Kuhlmann T, Antel J. The role of glial cells in multiple sclerosis disease progression. Nat Rev Neurol 2022;18:237–48, doi: 10.1038/s41582-022-00624-x.
86. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer 2014;14:219–32, doi: 10.1038/nrc3702.
87. Herman AE, et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC-0853, a selective reversible Bruton’s tyrosine kinase inhibitor. Clin Pharmacol Ther 2018;103:1020–8, doi: 10.1002/cpt.1056.
88. Herman SE, et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia 2014;28:2188–96, doi: 10.1038/leu.2014.122.
89. Horwood NJ, et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol 2006;176:3635–41, doi: 10.4049/jimmunol.176.6.3635.
90. Imbruvica (Ibrutinib) Kapseln und Tabletten [Fachinformation]. (Jansset Biotech, Inc., Horsham, PA, 2020).
91. Iwaki S, et al. Btk plays a crucial role in the amplification of FcεRI-mediated mast cell activation by Kit. J Biol Chem 2005;280:40261–70, doi: 10.1074/jbc.M506063200.
92. Jackle K, et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain 2020;143:2073–88, doi: 10.1093/brain/awaa158.
93. Jain RW, Yong VW. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol 2021;22:513–24, doi: 10.1038/s41577-021-00652-6.
94. Jefferies CA, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem 2003;278:26258–64, doi: 10.1074/jbc.M301484200.
95. Jeske SS, et al. Adenosine-producing regulatory B cells in head and neck cancer. Cancer Immunol Immunother 2020;69:1205–16, doi: 10.1007/s00262-020-02535-6.
96. Jongstra-Bilen J, et al. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol 2008;181:288–98, doi: 10.4049/jimmunol.181.1.288.
97. Kamma E, Lasisi W, Libner C, Ng HS, et al. Central nervous system macrophages in progressive multiple sclerosis: relationship to neurodegeneration and therapeutics. J Neuroinflammation 2022;19:45, doi: 10.1186/s12974-022-02408-y.
98. Kapseln Brukinsa (Zanubrutinib) [Fachinformation]. (BeiGene USA, Inc., San Mateo, CA, 2019).
99. Kaul M, et al. Remibrutinib (LOU064): a selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin Transl Sci 2021;14:1756–68, doi: 10.1111/cts.13005.
100. Kawakami Y, et al. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon Fc epsilon RI cross-linking. Mol Cell Biol 1994;14:5108–13, doi: 10.1128/mcb.14.8.5108-5113.1994.
101. Kenny EF, et al. Bruton’s tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS one 2013;8:e74103, doi: 10.1371/journal.pone.0074103.
102. Kiefer F, et al. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol Cell Biol 1998;18:4209–20, doi: 10.1128/mcb.18.7.4209.
103. Kil LP, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 2012;119:3744–3756, doi: 10.1182/blood-2011-12-397919.
104. Kiss R, et al. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg Med Chem Lett 2009;19:3598–601, doi: 10.1016/j.bmcl.2009.04.138.
105. Komori M, et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol 2016;3:166–79, doi: 10.1002/acn3.293.
106. Kowarik MC, et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation 2012;9:93, doi: 10.1186/1742-2094-9-93.
107. Krämer J, et al. Imaging in mice and men: pathophysiological insights into multiple sclerosis from conventional and advanced MRI techniques. Prog Neurobiol 2019;182:101663, doi: 10.1016/j.pneurobio.2019.101663.
108. Kuhle J, et al. Evobrutinib, a Bruton’s tyrosine kinase inhibitor, decreases neurofilament light chain levels over 2.5 years of treatment in patients with relapsing multiple sclerosis. Mult Scler 2022;28(3_suppl):EP1021.
109. Kuhlmann T, et al. An updated histological classification system for multiple sclerosis lesion. Acta Neuropathol 2017;133:13–24, doi: 10.1007/s00401-016-1653-y.
110. Lassmann H. The contribution of neuropathology to multiple sclerosis research. Eur J Neurol 2022;29:2869–77, doi: 10.1111/ene.15360.
111. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol 2018;9:3116, doi: 10.3389/fimmu.2018.03116.
112. Lassmann H, Raine CS, Antel J, Prineas JW. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol 1998;86:213–7, doi: 10.1016/s0165-5728(98)00031-9.
113. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov 2021;20:179–99, doi: 10.1038/s41573-020-00092-2.
114. Lee KG, et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci USA 2012;109:5791–6, doi: 10.1073/pnas.1119238109.
115. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 2018;18:225–42, doi: 10.1038/nri.2017.125.
116. Li R, et al. BTK inhibition limits B-cell–T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol 2022;143:505–21, doi: 10.1007/s00401-022-02411-w.
117. Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol 2018;19:696–707, doi: 10.1038/s41590-018-0135-x.
118. Liang C, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem 2018;151:315–26, doi: 10.1016/j.ejmech.2018.03.062.
119. Lin X, Wang D. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin Immunol 2004;16:429–35, doi: 10.1016/j.smim.2004.08.022.
120. Lipsky A, Lamanna N. Managing toxicities of Bruton tyrosine kinase inhibitors. Hematol Am Soc Hematol Educ Program 2020;2020;336–45, doi: 10.1182/hematology.2020000118.
121. Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal 1998;10:529–42, doi: 10.1016/s0898-6568(98)00012-6.
122. Liu X, et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase BTK. Nat Immunol 2011;12:416–24, doi: 10.1038/ni.2015.
123. Lloyd AF, Miron VE. The pro-remyelination properties of microglia in the central nervous system. Nat Rev Neurol 2019;15:447–58, doi: 10.1038/s41582-019-0184-2.
124. Luchetti S, et al. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol 2018;135:511–28, doi: 10.1007/s00401-018-1818-y.
125. Lunemann JD, Malhotra S, Shinohara ML, Montalban X, et al. Targeting inflammasomes to treat neurological disease. Ann Neurol 2021;90:177–88, doi: 10.1002/ana.26158.
126. Lünemann JD, Ruck T, Muraro PA, Bar-Or A, et al. Immune reconstitution therapies: concepts for durable remission in multiple sclerosis. Nat Rev Neurol 2020;16:56–62, doi: 10.1038/s41582-019-0268-z.
127. Luo C, et al. The role of microglia in multiple sclerosis. Neuropsychiatr Dis Treat 2017;13:1661–7, doi: 10.2147/ndt.S140634.
128. Magliozzi R, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089–104, doi: 10.1093/brain/awm038.
129. Mahajan S, et al. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton’s tyrosine kinase (BTK), LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2,5-dibromophenyl)propenamide]. J Biol Chem 1999;274:9587–99, doi: 10.1074/jbc.274.14.9587.
130. Mangla A, et al. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood 2004;104:1191–7, doi: 10.1182/blood-2004-01-0207.
131. Mano H. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev 1999;10:267–80, doi: 10.1016/s1359-6101(99)00019-2.
132. Martin E, et al. Bruton’s tyrosine kinase inhibition promotes myelin repair. Brain Plast 2020;5:123–33, doi: 10.3233/bpl-200100.
133. Medzhitov R, Janeway C, Jr. Innate immune recognition: mechanisms and pathway. Immunol Rev 2000;173:89–97, doi: 10.1034/j.1600-065x.2000.917309.x.
134. Middendorp S, Dingjan GM, Hendriks RW. Impaired precursor B cell differentiation in Bruton’s tyrosine kinase-deficient mice. J Immunol 2002;168:2695–703, doi: 10.4049/jimmunol.168.6.2695.
135. Minagar A, Maghzi AH, McGee JC, Alexander JS. Emerging roles of endothelial cells in multiple sclerosis pathophysiology and therapy. Neurol Res 2012;34:738–45, doi: 10.1179/1743132812y.0000000072.
136. Mohamed AJ, et al. Bruton’s tyrosine kinase (BTK): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 2009;228:58–73, doi: 10.1111/j.1600-065X.2008.00741.x.
137. Montalban X, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209–20, doi: 10.1056/NEJMoa1606468.
138. Montalban X, et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med 2019;380:2406–17, doi: 10.1056/NEJMoa1901981.
139. Montalban X, et al. Safety and efficacy of evobrutinib, a Bruton tyrosine kinase inhibitor, in relapsing multiple sclerosis over 2.5 years of the open-label extension to a phase 2 trial. Int J MS Care 2022;21(Suppl 1):DMT02.
140. Mrdjen D, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 2018;48:599, doi: 10.1016/j.immuni.2018.02.014.
141. Muroi M, Tanamoto K. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB. J Leukoc Biol 2008;83:702–7, doi: 10.1189/jlb.0907629.
142. Muta T, et al. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature 1994;368:70–3, doi: 10.1038/368070a0.
143. Netea MG, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020;20:375–88, doi: 10.1038/s41577-020-0285-6.
144. Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, et al. Bruton’s tyrosine kinase is essential for human B cell tolerance. J Exp Med 2004;200:927–34, doi: 10.1084/jem.20040920.
145. Nishizumi H, Horikawa K, Mlinaric-Rascan I, Yamamoto T. A double-edged kinase Lyn: a positive and negative regulator for antigen receptor-mediated signals. J Exp Med 1998;187:1343–8, doi: 10.1084/jem.187.8.1343.
146. Nyhoff LE, et al. Bruton’s tyrosine kinase is not essential for B cell survival beyond early developmental stage. J Immunol 2018;200:2352–61, doi: 10.4049/jimmunol.1701489.
147. Oh J, et al. MRI, safety, and efficacy outcomes in patients with relapsing MS: 18-month results from the long-term extension study of tolebrutinib. Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Forum (West Palm Beach, FL, USA) 102 (ACTRIMS, 2022).
148. Okada T, Maeda A, Iwamatsu A, Gotoh K, et al. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity 2000;13:817–27, doi: 10.1016/s1074-7613(00)00079-0.
149. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007;7:353–64, doi: 10.1038/nri2079.
150. O’Rourke LM, et al. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity 1998;8:635–45, doi: 10.1016/s1074-7613(00)80568-3.
151. Owens TD, et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin Transl Sci 2022;15:442–50, doi: 10.1111/cts.13162.
152. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer 2018;17:57, doi: 10.1186/s12943-018-0779-z.
153. Pan Z, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007;2:58–61, doi: 10.1002/cmdc.200600221.
154. Pellerin K, et al. MOG autoantibodies trigger a tightly-controlled FcR and BTK-driven microglia proliferative response. Brain 2021;144:2361–74, doi: 10.1093/brain/awab231.
155. Peng SL, Gerth AJ, Ranger AM, Glimcher LH. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 2001;14:13–20, doi: 10.1016/s1074-7613(01)00085-1.
156. Piasecka-Stryczynska K, et al. Concentration of evobrutinib, a BTK inhibitor, in cerebrospinal fluid during treatment of patients with relapsing multiple sclerosis in a phase 2 study. Mult Scler Relat Disord 2021;51:103001, doi: 10.1016/j.msard.2021.103001.
157. Preziosa P, et al. Slowly expanding lesions predict 9-year multiple sclerosis disease progression. Neurol Neuroimmunol Neuroinflamm 2022;9:e1139, doi: 10.1212/nxi.0000000000001139.
158. Puthenparampil M, et al. BAFF index and CXCL13 levels in the cerebrospinal fluid associate respectively with intrathecal IgG synthesis and cortical atrophy in multiple sclerosis at clinical onset. J Neuroinflammation 2017;14:11, doi: 10.1186/s12974-016-0785-2.
159. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 2009;27:119–45, doi: 10.1146/annurev.immunol.021908.132528.
160. Rawlings DJ, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science 1993;261:358–61, doi: 10.1126/science.8332901.
161. Reich DS, et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2021;20:729–38, doi: 10.1016/S1474-4422(21)00237-4.
162. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med 2018;378:169–80, doi: 10.1056/NEJMra1401483.
163. Rijvers L, et al. Human T-bet + B cell development is associated with BTK activity and suppressed by evobrutinib. JCI Insight 2022;7, doi: 10.1172/jci.insight.160909.
164. Ringheim GE, Wampole M, Oberoi K. Bruton’s tyrosine kinase (BTK) inhibitors and autoimmune diseases: making sense of BTK inhibitor specificity profiles and recent clinical trial successes and failures. Front Immunol 2021;12:662223, doi: 10.3389/fimmu.2021.662223.
165. Rip J, et al. Toll-like receptor signaling drives BTK-mediated autoimmune disease. Front Immunol 2019;10:95, doi: 10.3389/fimmu.2019.00095.
166. Rip J, Van Der Ploeg EK, Hendriks RW, Corneth OBJ. The role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit Rev Immunol 2018;38:17–62, doi: 10.1615/CritRevImmunol.2018025184.
167. Rotstein DL. All Bruton’s tyrosine kinase inhibitors have similar efficacy and risks: no. Mult Scler 2022;28:1500–2, doi: 10.1177/13524585221091060.
168. Rowley RB, Burkhardt AL, Chao HG, Matsueda GR, et al. Syk protein-tyrosine kinase is regulated by tyrosine-phosphorylated Ig alpha/Ig beta immunoreceptor tyrosine activation motif binding and autophosphorylation. J Biol Chem 1995;270:11590–4, doi: 10.1074/jbc.270.19.11590.
169. Ruck T, Nimmerjahn F, Wiendl H, Lünemann JD. Next-generation antibody-based therapies in neurology. Brain 2022;145:1229–41, doi: 10.1093/brain/awab465.
170. Ruiz F, Vigne S, Pot C. Resolution of inflammation during multiple sclerosis. Semin Immunopathol 2019;41:711–26, doi: 10.1007/s00281-019-00765-0.
171. Saito K, Scharenberg AM, Kinet JP. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates BTK. J Biol Chem 2001;276:16201–6, doi: 10.1074/jbc.M100873200.
172. Saitoh S, et al. LAT is essential for Fc(epsilon)RI-mediated mast cell activation. Immunity 2000;12:525–35, doi: 10.1016/s1074-7613(00)80204-6.
173. Schaeffer EM, et al. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science 1999;284:638–41, doi: 10.1126/science.284.5414.638.
174. Schmitz R, Baumann G, Gram H. Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J Mol Biol 1996;260:664–77, doi: 10.1006/jmbi.1996.0429.
175. Shinohara H, Maeda S, Watarai H, Kurosaki T. IkappaB kinase beta-induced phosphorylation of CARMA1 contributes to CARMA1 Bcl10 MALT1 complex formation in B cells. J Exp Med 2007;204:3285–93, doi: 10.1084/jem.20070379.
176. Simmons SB, Ontaneda D. Slowly expanding lesions: a new target for progressive multiple sclerosis trials. Neurology 2022;98:699–700, doi: 10.1212/wnl.0000000000200230.
177. Simon M, Vanes L, Geahlen RL, Tybulewicz VL. Distinct roles for the linker region tyrosines of Syk in FcepsilonRI signaling in primary mast cells. J Biol Chem 2005;280:4510–7, doi: 10.1074/jbc.M410326200.
178. Smith CI, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol 1994;152:557–65.
179. Smith CI, et al. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 2001;23:436–46, doi: 10.1002/bies.1062.
180. Smith P, et al. Phase 1 clinical trial of PRN2246 (SAR441268), a covalent BTK inhibitor demonstrates safety, CNS exposure and therapeutic levels of BTK occupancy. Mult Scler 2019;25(Suppl_1):P072.
181. Solvason N, et al. Transgene expression of bcl-xL permits anti-immunoglobulin (Ig)-induced proliferation in xid B cells. J Exp Med 1998;187:1081–91, doi: 10.1084/jem.187.7.1081.
182. Spaargaren M, et al. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med 2003;198:1539–50, doi: 10.1084/jem.20011866.
183. Steinmaurer A, Wimmer I, Berger T, Rommer PS, et al. Bruton’s tyrosine kinase inhibition in the treatment of preclinical models and multiple sclerosis. Curr Pharm Des 2022;28:437–44, doi: 10.2174/1381612827666210701152934.
184. Su YW, et al. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur J Immunol 1999;29:3702–11, doi: 10.1002/(sici)1521-4141(199911)29:11<3702::Aid-immu3702>3.0.Co;2-r.
185. Sucksdorff M, et al. Brain TSPO-PET predicts later disease progression independent of relapses in multiple sclerosis. Brain 2020;143:3318–30, doi: 10.1093/brain/awaa275.
186. Syed S, et al. Efficacy and safety of tolebrutinib in patients with highly active relapsing MS: subgroup analysis of the phase 2b study. Neurology 2021;96(Suppl 15):2260.
187. Tam CS, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenstrom macroglobulinemia: the ASPEN study. Blood 2020;136:2038–50, doi: 10.1182/blood.2020006844.
188. Tanwar S, et al. Mediation of transitional B cell maturation in the absence of functional Bruton’s tyrosine kinase. Sci Rep 2017;7:46029, doi: 10.1038/srep46029.
189. Taylor CW, Tovey SC. IP(3) receptors: toward understanding their activation. Cold Spring Harb Perspect Biol 2010;2:a004010, doi: 10.1101/cshperspect.a004010.
190. Teixeira C, Stang SL, Zheng Y, Beswick NS, et al. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood 2003;102:1414–20, doi: 10.1182/blood-2002-11-3621.
191. Thomas JD, et al. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 1993;261:355–8, doi: 10.1126/science.8332900.
192. Timofeeva N, Gandhi V. Ibrutinib combinations in CLL therapy: scientific rationale and clinical results. Blood Cancer J 2021;11:79, doi: 10.1038/s41408-021-00467-7.
193. Torke S, Weber MS. Inhibition of Bruton’s tyrosine kinase as a novel therapeutic approach in multiple sclerosis. Exp Opin Investig Drugs 2020;29:1143–50, doi: 10.1080/13543784.2020.1807934.
194. Torke S, et al. Inhibition of Bruton’s tyrosine kinase interferes with pathogenic B-cell development in inflammatory CNS demyelinating disease. Acta Neuropathol 2020;140:535–48, doi: 10.1007/s00401-020-02204-z.
195. Tsouki F, Williams A. Multifaceted involvement of microglia in gray matter pathology in multiple sclerosis. Stem Cell 2021;39:993–1007, doi: 10.1002/stem.3374.
196. Tsukada S, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993;72:279–90, doi: 10.1016/0092-8674(93)90667-f.
197. Turner T, Brun P, Ofengheim D, Gruber R. Comparative CNS pharmacology of tolebrutinib versus other BTK inhibitor candidates for treating MS. Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Forum (West Palm Beach, FL, USA) 162 (ACTRIMS, 2022).
198. Uckun FM, et al. BTK as a mediator of radiation-induced apoptosis in DT-40 lymphoma B cells. Science 1996;273:1096–100, doi: 10.1126/science.273.5278.1096.
199. Väliaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat 2006;27:1209–17, doi: 10.1002/humu.20410.
200. van den Akker E, et al. The Btk inhibitor LFM-A13 is a potent inhibitor of Jak2 kinase activity. Biol Chem 2004;385:409–13, doi: 10.1515/bc.2004.045.
201. Vermersch P, et al. MRI and clinical outcomes of evobrutinib, a Bruton’s tyrosine kinase inhibitor, in relapsing multiple sclerosis over 2.5 years of the open-label extension to a phase 2 trial. Mult Scler 2022;28(3_suppl):P731.
202. Vetrie D, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993;361:226–33, doi: 10.1038/361226a0.
203. Wang J, et al. Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front Pharmacol 2019;10:286, doi: 10.3389/fphar.2019.00286.
204. Weber M, et al. Fenebrutinib reduces disease activity in a mouse model of infammatory multiple sclerosis, which is associated with reduced microglial activation. Mult Scler 2021;27(Suppl_2):P680.
205. Weber M, et al. Fenebrutinib demonstrates the highest potency of Bruton tyrosine kinase inhibitors (BTKis) in phase 3 clinical development for multiple sclerosis (MS). Neurology 2021;96(Suppl 15):4437.
206. Wen T, Wang J, Shi Y, Qian H, et al. Inhibitors targeting Bruton’s tyrosine kinase in cancers: drug development advances. Leukemia 2021;35:312–32, doi: 10.1038/s41375-020-01072-6.
207. Westerberg L, Greicius G, Snapper SB, Aspenström P, et al. Cdc42, Rac1, and the Wiskott–Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood 2001;98:1086–94, doi: 10.1182/blood.v98.4.1086.
208. Xu Y, Fairfax K, Light A, Huntington ND, et al. CD19 differentially regulates BCR signalling through the recruitment of PI3K. Autoimmunity 2014;47:430–7, doi: 10.3109/08916934.2014.921810.
209. Yang EJ, Yoon JH, Chung KC. Bruton’s tyrosine kinase phosphorylates cAMP-responsive element-binding protein at serine 133 during neuronal differentiation in immortalized hippocampal progenitor cells. J Biol Chem 2004;279:1827–37, doi: 10.1074/jbc.M308722200.
210. Yasuda T, et al. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity 2008;28:499–508, doi: 10.1016/j.immuni.2008.02.015.
211. Ying H, Li Z, Yang L, Zhang J. Syk mediates BCR- and CD40-signaling integration during B cell activation. Immunobiology 2011;216:566–70, doi: 10.1016/j.imbio.2010.09.016.
212. Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol 2022;18:40–55, doi: 10.1038/s41582-021-00581-x.
213. Yu CG, et al. Inhibition of Bruton tyrosine kinase reduces neuroimmune cascade and promotes recovery after spinal cord injury. Int J Mol Sci 2021;23, doi: 10.3390/ijms23010355.
214. Zain R, Vihinen M. Structure–function relationships of covalent and non-covalent BTK inhibitors. Front Immunol 2021;12:694853, doi: 10.3389/fimmu.2021.694853.
215. Zhang B, et al. Orelabrutinib, a potent and selective Bruton’s tyrosine kinase inhibitor with superior safety profile and excellent PK/PD properties. Cancer Res 2020;80(Suppl 16):CT132.
Dr. med. Julia Krämer, Prof. Dr. med. Heinz Wiendl, Klinik für Neurologie mit Institut für Translationale Neurologie, Universitätsklinikum Münster, Albert-Schweitzer-Campus 1, Gebäude A1, D-48149 Münster, Deutschland, E-Mail: julia.kraemer@ukmuenster.de (JK); wiendl@uni-muenster.de (HW)
Bruton tyrosine kinase inhibitors for multiple sclerosis
Current therapies for multiple sclerosis (MS) effectively reduce relapses and relapse-associated worsening assumed to be mainly associated with transient infiltration of peripheral immune cells into the central nervous system (CNS). However, they are less effective at slowing disability accumulation in MS, likely reflecting in part their lack of relevant effects on CNS-compartmentalized inflammation.
Bruton’s tyrosine kinase (BTK) is an intracellular signalling molecule regulating maturation, survival, migration, and activation of B cells and microglia. CNS-compartmentalized B cells and microglia are considered central players in immunopathogenesis of progressive disease mechanisms; accordingly, CNS-penetrant inhibitors of BTK (BTKis) hold promise to curtail disease progression by targeting immune cells on both sides of the blood-brain barrier.
Originally developed as a B-cell malignancy treatment, currently five BTKis are undergoing clinical trials in MS. These BTKis differ in selectivity, strength of BTK inhibition, binding mechanisms, and ability to modulate immune cells within the CNS.
Here, we review BTK function in signalling pathways of different immune cells relevant to MS, provide an overview of preclinical data, and discuss clinical trial results.
Key words: BTK inhibitors, multiple sclerosis, B cells, microglia, disease progression
Psychopharmakotherapie 2023; 30(05):146-163