Gerrit Machetanz*, Andreas Wolff* und Paul Lingor
Die kontrovers diskutierte Zulassung von Aducanumab für die Behandlung der Alzheimer-Erkrankung (engl. Alzheimer’s disease, AD) durch die Food and Drug Administration (FDA) hat 2021 ein Schlaglicht auf die Entwicklung von Antikörper-Therapien bei neurodegenerativen Erkrankungen geworfen [55]. Auch für den Morbus Parkinson (engl. Parkinson’s disease, PD) sind Antikörper-basierte Immuntherapien in Entwicklung, die wir in dem folgenden Artikel diskutieren wollen.
Morbus Parkinson ist nach die Alzheimer-Erkrankung die zweithäufigste neurodegenerative Erkrankung. Inzidenz sowie Prävalenz nehmen in Europa und weltweit weiter zu [15]. Dabei ist diese Zunahme durch eine alternde Bevölkerung und eine akkuratere Diagnosestellung nur teilweise erklärt [5]. Neben der ausgeprägten Belastung, die die Erkrankung für Betroffene und Angehörige mit sich bringt, hat Morbus Parkinson somit auch weiter zunehmende sozioökonomische Bedeutung [25]. Neuropathologisch wird Morbus Parkinson unter anderem durch den Verlust dopaminerger Neurone in der Substantia nigra pars compacta (SNpc) sowie durch vornehmlich aus Alpha-Synuclein (aSyn) bestehende Proteinaggregate – Lewy-Körperchen und Lewy-Neuriten – charakterisiert, wobei die Pathologie deutlich darüber hinaus geht und zahlreiche andere Bereiche des zentralen Nervensystems (ZNS) betrifft [16, 67].
Gegenwärtige Therapien sind vor allem in frühen Phasen der Erkrankung sehr gut symptomatisch wirksam. Sie haben jedoch keinen Einfluss auf die Progression: Weder der Verlust weiterer Neurone noch das Fortschreiten der Lewy-Pathologie werden verzögert oder gar rückgängig gemacht. Es bedarf also dringend neuer, krankheitsmodifizierender therapeutischer Möglichkeiten. Einer der gegenwärtig untersuchten Ansätze sind Immuntherapien, mit deren Hilfe die Bildung, die extrazelluläre Freisetzung und/oder neuronale Transmission pathologischer aSyn-Aggregate verhindert werden sollen [26]. Der biologische Hintergrund und der gegenwärtige Stand der Entwicklung von Antikörper-basierten Immuntherapien bei Morbus Parkinson sollen hier dargestellt werden.
Ansatzpunkte für krankheitsmodifizierende Therapien
Beim weitaus größten Teil der Patienten mit Morbus Parkinson kennen wir keine monogenetische Ursache der Erkrankung und sprechen daher von einem sporadischen Auftreten. Mutationen und Varianten in zahlreichen Genen haben aber wichtige Erkenntnisse über die Pathophysiologie der Erkrankung geliefert. So können Punktmutationen, Duplikationen und Triplikationen im für aSyn kodierenden SNCA-Gen Morbus Parkinson verursachen und in genomweiten Assoziationsstudien (GWAS) zeigt der SNCA-Locus eine starke Assoziation mit der Erkrankung [9, 54, 65, 66]. Lewy-Körperchen und Lewy-Neuriten bestehen vor allem aus aSyn-Aggregaten, daher wird der Aggregation von aSyn eine zentrale Rolle in der Entstehung von Morbus Parkinson zugesprochen [67].
Mutationen in anderen Genen können orientierend in Gruppen eingeteilt werden, die eine Dysfunktion der Mitochondrien (PRKN, PINK1, DJ-1, FBXO7) oder aber der Lysosomen und des Vesikeltransports (GBA, LRRK2, VPS35) zur Folge haben. Es wird angenommen, dass diese Fehlfunktionen im gleichen Endpunkt, der aSyn-Aggregation und dem Verlust dopaminerger Neurone, münden und dass Dysfunktionen in diesen Regelkreisläufen auch in der Pathogenese des sporadischen Morbus Parkinson eine entscheidende Rolle spielen [4]. Dabei findet sich jedoch bei einigen Patienten mit einer monogenetischen Parkinsonerkrankung nicht immer eine aSyn-Pathologie [62].
In seiner physiologischen Form ist aSyn im ZNS am ehesten als ungefaltetes Monomer oder Alpha-helikales Tetramer vor allem in präsynaptischen Endigungen von Neuronen lokalisiert. Es wird eine Rolle in der Neurotransmitter-Ausschüttung postuliert, insbesondere der Bildung von SNARE(soluble N-ethylmaleimide-sensitive-factor attachment receptor)-Komplexen und der synaptischen Plastizität. Seine genaue Funktion wird aber bis heute nicht ausreichend verstanden [69].
Im pathologischen Prozess wird monomeres aSyn in eine β-Faltblattstruktur konvertiert, die sich weiter als toxische Oligomere und Fibrillen organisieren kann. Verschiedene Mechanismen können diese Fehlfaltung initiieren: Überexpression von aSyn [54, 66], niedriger pH-Wert [44], Überproduktion von reaktiven Sauerstoffspezies (ROS) und Störung der Mitochondrienfunktion [64, 72], Interaktion mit Lipiden und Zellmembranen [70] sowie die Dysregulation von Protein-Abbauprozessen [10, 75].
Darüber hinaus sind inzwischen verschiedene aSyn-„Stämme“ identifiziert worden, die sich durch die strukturell unterschiedliche Zusammensetzung der aSyn-Fibrillen definieren. Es wird diskutiert, dass die verschiedenen Stämme
- für die unterschiedlichen Pathologien und klinischen Verläufe der neben Morbus Parkinson existierenden, distinkten Synucleinopathien (Multisystematrophie [MSA], Demenz mit Lewy-Körperchen [DLB], reines autonomes Versagen [pure autonomic failure, PAF])
- und für interindividuell unterschiedliche Krankheitsverläufe bei Patienten innerhalb der jeweiligen Erkrankung
mitverantwortlich sind [37].
Posttranslationale Modifikationen können Struktur, Funktion, Abbau und Lokalisation von Proteinen und so auch von aSyn modulieren. Die häufigsten posttranslationalen Modifikationen von aSyn im Hirngewebe von Betroffenen mit Morbus Parkinson sind N-terminale Acetylierung, Phosphorylierung, Ubiquitinierung, Nitrierung und Trunkierung. Allerdings ist gegenwärtig nicht ausreichend geklärt, wie posttranslationale Modifikationen die strukturellen Eigenschaften von aSyn-Oligomeren und -Fibrillen sowie deren Toxizität konkret beeinflussen [49].
Aggregiertes aSyn kann in den Extrazellularraum freigesetzt werden, weiter in andere Neuronen und Gliazellen gelangen und dort die weitere Fehlfaltung und Aggregation von monomerem aSyn initiieren [26]. Dieser Vorgang wird als „seeding“ oder „permissive templating“ bezeichnet und wurde bei Patienten mit Morbus Parkinson vermutet, die in Therapiestudien eine Transplantation fetaler nigraler Zellen erhalten hatten und bei denen nach wenigen Jahren im transplantierten Gewebe Lewy-Körperchen nachgewiesen wurden. In zahlreichen Tiermodellen konnte dieser Seeding-Mechanismus ebenfalls nachvollzogen werden [12, 33, 35]. Ein indirekter Hinweis für das „permissive templating“ sind die Braak-Stadien. Sie beschreiben bei Patienten mit Morbus Parkinson die topographisch einem sequenziellen Muster folgende Ausbreitung der aSyn-Pathologie in verschiedene Regionen des peripheren und zentralen Nervensystems und sind gut mit einer Ausbreitung der Pathologie von Zelle zu Zelle vereinbar [6].
Die Freisetzung von aggregiertem aSyn in den Extrazellularraum erfolgt durch klassische, ER(endoplasmatisches Retikulum)-Golgi-Apparat-abhängige und nichtklassische, ER-Golgi-Apparat-unabhängige Exozytose oder vesikelassoziiert, beispielsweise in Form von Exosomen. Eine Zell-zu-Zell-Transmission kann auch über „tunneling nanotubes“ stattfinden [23, 24]. Vom Extrazellularraum ausgehend bindet aggregiertes aSyn an die gesamte Zelloberfläche von Neuronen. Die Aufnahme in die Zelle funktioniert über die Interaktion mit verschiedenen transmembranösen Proteinen und Rezeptoren. Die Blockade der Aufnahme von toxischen aSyn-Spezies in die Zelle könnte theoretisch auch ein Fortschreiten der Pathologie verhindern. Daher erscheinen die bisher unter anderem als Mediatoren der Aufnahme identifizierten Heparansulfatproteoglykane (HSPG) LAG3 (Lymphocyte-activation gene 3), Neurexin1β und CX32 (Connexin 32) auch als potenzielle Therapieziele [3, 27, 41, 56].
Aggregiertes aSyn entfaltet seine toxische Wirkung über eine Reihe von Mechanismen. Es stört unter anderem die Bildung von SNARE-Komplexen, reduziert die Beweglichkeit synaptischer Vesikel, fördert mitochondriale Dysfunktion, beeinträchtigt den Transport von Proteinen vom ER zum Golgi-Apparat (in diesem Rahmen auch die Autophagie) und führt außerdem zu lysosomaler Dysfunktion [75].
In der Zusammenschau scheint aSyn ein zentraler Faktor in der PD-Pathogenese und somit auch ein attraktiver direkter Angriffspunkt für Immuntherapien zu sein (Abb. 1).
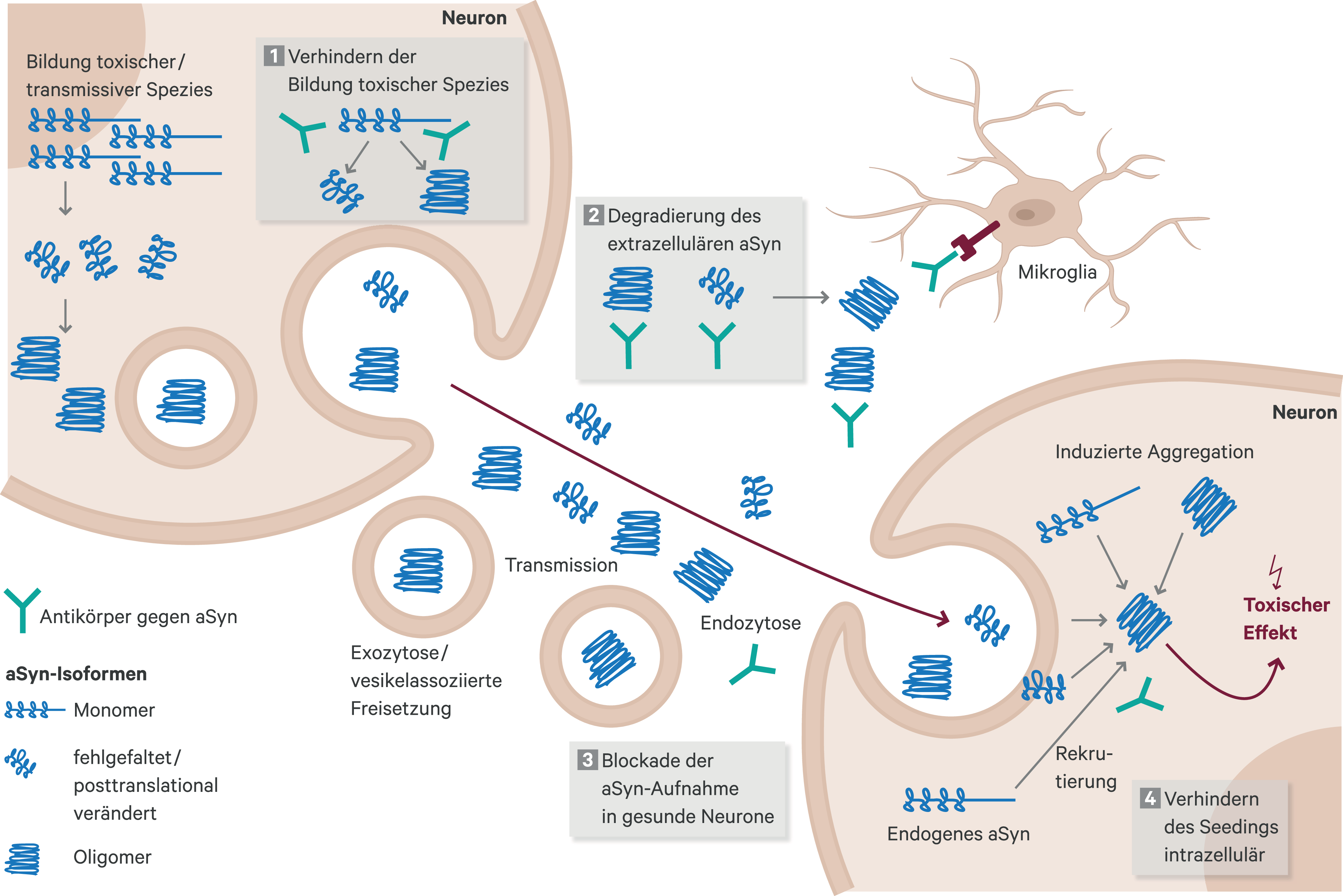
Abb. 1. Mögliche zelluläre und molekulare Angriffspunkte einer Antikörper-basierten Immuntherapie gegen M. Parkinson Alpha-Synuclein (aSyn) wird fehlgefaltet und organisiert sich in Oligomeren, die dann mittels Exozytose oder vesikelassoziierter Exozytose in den Extrazellularraum gelangen. Von dort dringt das aggregierte aSyn unter anderem mittels Endozytose in gesunde Neurone ein, wo es aSyn-Monomere rekrutiert und die weitere Fehlfaltung initiiert (sog. seeding oder permissive templating). Mögliche Angriffspunkte einer Immuntherapie in diesem Prozess könnten sein: 1.) Verhinderung der aSyn-Aggregation und somit der Bildung potenziell toxischer aSyn-Spezies, 2.) Bindung von Antikörpern an aggregiertes aSyn im Extrazellularraum und Zuführung der Komplexe zum Abbau in Mikroglia, 3.) Blockade der Aufnahme von toxischen aSyn-Spezies in gesunde Neurone, 4.) Verhinderung der Rekrutierung von monomerem aSyn und der weiteren Entstehung von intrazellulär aggregiertem aSyn in gesunden Neuronen.
Es muss dabei jedoch auch erwähnt werden, dass durchaus kritisch diskutiert wird, ob die Aggregation von aSyn in jedem Fall pathogen ist, oder ob sie möglicherweise auch ein Epiphänomen darstellen oder gar protektiv sein könnte. Dabei wird die Aggregation
- als gemeinsame Endstrecke verschiedener vorgeschalteter pathogener Prozesse ohne eigene krankheitsverursachende Wirkung oder gar
- als protektiver Mechanismus bei zellulärem Stress eingeordnet [19, 68].
Gegen aggregiertes aSyn gerichtete Therapien würden in diesem Fall keinen oder sogar einen negativen Effekt auf das Fortschreiten der Erkrankung haben.
Gerade deshalb stellen andere Ansatzpunkte, beispielsweise eine Verbesserung der mitochondrialen oder lysosomalen Funktion, valide alternative Therapieziele für einen krankheitsmodifizierenden Effekt dar.
Antikörper-basierte Immuntherapien
Als mögliche verlaufsmodifizierende Therapie wurde in den letzten zwei Jahrzehnten unter anderem die Immuntherapie untersucht, um die Ausbreitung der aSyn-Pathologie im Gehirn Erkrankter positiv zu beeinflussen oder gar zu verhindern. Antikörper (AK) könnten in die Bildung potenziell toxischer aSyn-Spezies eingreifen, bereits vorhandene pathologische Formen opsonieren oder toxische aSyn-Spezies abschirmen und somit zu einer Krankheitsmodifikation beitragen (Abb. 1). Sie könnten letztlich eine Verringerung von aSyn im extrazellulären Raum zur Folge haben und sich somit positiv auf das Fortschreiten von Morbus Parkinson auswirken [34]. Grundsätzlich können zwei Arten immuntherapeutischer Ansätze unterschieden werden: aktive Immunisierung und passive Antikörper-Therapie (Tab. 1). Bei der aktiven Immunisierung wird bei Patienten mittels eines Vakzins eine Immunantwort ausgelöst, die zur Produktion spezifischer, gegen das Zielprotein gerichteter Antikörper führt. Bei der passiven Immuntherapie hingegen werden vorgefertigte Antikörper verabreicht, die an das Zielprotein binden. Monoklonale – also aus einem Zellklon generierte – Antikörper (mAK) finden seit Längerem Einsatz in der Behandlung von Krebs- und Autoimmunerkrankungen und werden derzeit zur Behandlung von COVID-19 erprobt [30]. Zudem konnten bereits zur Jahrtausendwende erste Studien zur Immunisierung gegen Beta-Amyloid (Aβ), ein pathophysiologisch entscheidendes Protein bei der Entwicklung der Alzheimer-Erkrankung, zeigen, dass auch Proteinopathien aus dem Bereich der Neurodegeneration ein mögliches Anwendungsfeld für Antikörper-Therapien darstellen [14, 17]. Nachdem die Immunisierung gegen Aβ42 (unter dem Namen AN 1792) und dem Adjuvans QS-21, einem Extrakt aus dem chilenischen Seifenrindenbaum Quillaja saponaria, ein vielversprechendes Sicherheits- und Verträglichkeitsprofil in zwei Phase-I-Studien zeigte [1, 58], musste die nachfolgende Phase-II-Studie nach Auftreten von subakuten Meningoenzephalitiden bei 18 der 298 behandelten Patienten vorzeitig abgebrochen werden [50]. Fast 20 Jahre nach diesen ersten Versuchen der aktiven Immunisierung von an AD Erkrankten steht aktuell ein anderes Immuntherapeutikum gegen Beta-Amyloid, Aducanumab, wieder im Fokus. Nach widersprüchlichen Ergebnissen bezüglich der Wirksamkeit zur Behandlung demenzbezogener Symptome (gemessen mittels Clinical Dementia Rating Scale) [32] und Häufungen von sogenannten Amyloid-related imaging abnormalities, also bildgebenden Veränderungen bei behandelten Patienten in 35 % der Fälle, die bei 26 % symptomatisch wurden [57], lehnte die europäische Arzneimittelbehörde EMA die Zulassung für Europa ab, obwohl in den USA durch die FDA eine eingeschränkte Zulassung erfolgte. Zeitgleich wurden auch Anwendungsmöglichkeiten der Immuntherapie bei Synucleinopathien untersucht. Erste präklinische Daten lieferten Masliah et al., die bereits 2005 in einem transgenen Mausmodell histopathologisch positive Effekte durch eine Immunisierung mit humanem aSyn auf die Aggregation von aSyn zeigen konnten [42]. In den darauffolgenden Jahren konnte in Folgestudien gezeigt werden, dass auch monoklonale Antikörper gegen aSyn geeignet sind, phänotypische und neuropathologische Veränderungen in transgenen Mausmodellen zu reduzieren [43]. Grundsätzlich gilt es zu beachten, dass eine aktive Immunisierung im Vergleich zur passiven Immuntherapie möglicherweise sowohl kostengünstiger als auch weniger zeitaufwendig wäre, da die Wirkdauer nicht an die Halbwertszeit der verabreichten Antikörper gebunden ist. Eine passive Immunisierung erfordert eine wiederholte Verabreichung von ex vivo hergestellten, gut charakterisierten Antikörpern. Andererseits bieten die Messbarkeit der Antikörper-Serumspiegel und die begrenzten Halbwertszeiten der Antikörper im Körper auch die Möglichkeit, Nebenwirkungen zu kontrollieren und die Behandlung individuell anzupassen [2]. Ein Hauptproblem bei der Entwicklung von Antikörper-Therapien gegen neurodegenerative Erkrankungen stellte die pharmakokinetische Verteilung der Antikörper über die Blut-Hirn-Schranke (BHS) dar. Es ist davon auszugehen, dass nur 0,1 % bis 1,0 % der peripheren Antikörper die BHS überwinden [61]. Allerdings können monoklonale Antikörper molekularbiologisch verändert werden, um so den Übertritt über die BHS zu verbessern. Die Aufnahme von monoklonalen Antikörper über die BHS kann beispielsweise um den Faktor 80 erhöht werden, wenn diese an zwei einkettige variable Fragmente des Transferrin-Rezeptors gebunden werden [28].
Tab. 1. Ausgewählte aktuelle klinische Studien zu Antikörper-basierten Immuntherapien für Morbus Parkinson
|
Agens |
Sponsor und Studiennummer (ClinicalTrials.gov) |
Struktur und Target |
Studienphase |
Bisherige Ergebnisse |
|
|
Aktive Immunisierung |
AFFITOPE® PD01A |
AffiRIS AG NCT01568099 NCT01885494 NCT02216188 NCT02618941 NCT02270489* NCT02758730§ |
|
Phase I Abgeschlossen |
|
|
AFFITOPE® PD03A |
AffiRIS AG NCT02267434 NCT02270489* |
|
Phase I Abgeschlossen |
|
|
|
UB-312 |
United Neuroscience Ltd. NCT04075318 |
|
Phase I In Rekrutierung |
|
|
|
Passive Immunisierung |
Prasinezumab (PRX002) |
Hoffmann-La Roche/Prothena NCT02095171 NCT02157714 NCT03100149 NCT04777331 |
|
Phase II (PASADENA) Aktiv, nicht rekrutierend Phase IIb (PADOVA) In Rekrutierung |
|
|
Cinpanemab (BIIB054) |
Biogen NCT02459886 NCT03318523 NCT03716570 |
|
Phase II (SPARK) Vorzeitig abgebrochen |
|
|
|
MEDI1341 |
AstraZeneca NCT03272165 NCT04449484 |
|
Phase I bei Gesunden abgeschlossen |
|
|
|
Lu AF82 422 |
Lundbeck NCT03611569 NCT05104476* |
|
Phase I bei Gesunden und PD-Patienten abgeschlossen Phase II bei MSA-Patienten rekrutierend (AMULET) |
|
|
|
ABBV-0805 |
AbbVie NCT04127695§ |
|
Zurückgezogen |
|
*: bei MSA Patienten, §: zurückgezogen AK: Antikörper, aSyn: Alpha-Synuclein, NW: Nebenwirkung, mAK: monoklonaler Antikörper, MDS-UPDRS: Movement Disorder Society-Unified Parkinson‘s Disease Rating Scale, MSA: Multisystematrophie, PD: Morbus Parkinson
Aktive Immunisierung
Anfänglich erfolgten präklinische Versuche der aktiven Immuntherapie für Synucleinopathien mit dem aSyn-Protein in seiner vollen Länge [42]. Auch wenn die Ergebnisse dieser Untersuchungen vielversprechend waren, besteht bei der Immunisierung gegen das Gesamtprotein die Gefahr einer aSyn-spezifischen Autoimmunantwort. In Folgestudien wurde daher vor allem auf kürzere Epitope gesetzt, die eine ähnliche Antikörper-Antwort induzieren können, aber zu kurz für eine Aktivierung von zellulärer, T-Zell-vermittelter Autoimmunität sind [36, 61]. Zusätzlich gab es Versuche, die Immunogenität weiter zu optimieren. Hierzu wurden aSyn-Fragmente beispielsweise mit einem Epitop des Tetanustoxins (P30) [22] oder mit „virus-like particles“ (VLPs) [18] fusioniert oder in „glucan particles“, kleinen Zellmembransphären aus der Bäckerhefe, verpackt. Es kam jeweils zu einer robusten Immunantwort mit hohen Antikörper-Titern, die allerdings nicht immer zu einer Verbesserung der Symptomatik im untersuchten Mausmodell führte. Neben proteinbasierten Immunisierungsstudien wurde in den letzten Jahren auch die Entwicklung DNA- [11, 13, 31] oder Zell-basierter [71] Vakzine präklinisch erprobt. Mit beiden Systemen konnte eine Verbesserung der motorischen Endpunkte in vivo gezeigt werden, allerdings wurde diese bisher nur in präklinischen Studien untersucht. In der klinischen Entwicklung am weitesten fortgeschritten sind bis dato die AFFITOPE®-Vakzine der Firma AFFiRiS [60]. Hierbei handelt es sich um kurze Peptide mit einer im Vergleich zum ursprünglichen Epitop angepassten Aminosäuresequenz, die zu kurz sind, um eine T-Zell-Antwort auszulösen. Hierdurch wird es möglich, eine hohe Antikörper-Antwort zu erzeugen, während Kreuzreaktionen, beispielsweise mit Beta-Synuclein, vermieden werden können. In präklinischen Studien konnte hier in Mausmodellen für die Multisystematrophie, Demenz mit Lewy-Körperchen und Morbus Parkinson mit nachweislich positiven Effekten auf die Akkumulation von aSyn und motorische sowie kognitive Endpunkte ein „proof of concept“ geliefert werden [38–40]. Klinisch wurden die zwei AFFITOPE® PD01A und PD03A untersucht. Sie wiesen in Phase-I-Studien bei Patienten mit Multisystematrophie [45] und Morbus Parkinson [53, 73] ein günstiges Sicherheits- und Verträglichkeitsprofil über den Beobachtungszeitraum von 52 Wochen auf. Allerdings werden in Zukunft weitere Studien nötig sein, um die klinische Wirksamkeit zu evaluieren. Eine Phase-I-Studie zur Untersuchung von PD01A in PD-Patienten mit einer Mutation im GBA-Gen, dem häufigsten genetischen Risikofaktor für die Entwicklung einer Parkinson Erkrankung [2a], das das lysosomale Enzym Glucocerebrosidase kodiert, wurde vor Beginn der Rekrutierung zurückgezogen (NCT02758730). Zudem wird, nach vielversprechenden präklinischen Daten, aktuell ein weiteres aSyn-Vakzin klinisch evaluiert: Antikörper, die durch das auf einem zehn Aminosäuren langen, C-terminalen aSyn-Fragment basierende Vakzin UB-312 induziert wurden, erkannten die aSyn-Pathologie besser als kommerzielle Antikörper [47], schützten vor Verschlechterung der motorischen Funktion und reduzierten die Anreicherung von oligomerem aSyn in Gehirn und Darm eines PD-Mausmodells [46]. Seit August 2019 wird UB-312 in einer single-center Phase-I-Studie an gesunden Probanden und Patienten mit frühem Morbus Parkinson untersucht (NCT04075318).
Passive Antikörpertherapie
In verschiedenen PD-Mausmodellen führte die Behandlung mit dem monoklonalen AK 9E4 zu einer Reduktion der aSyn-Akkumulation und zu einer Besserung des motorischen und behavioralen Phänotyps [21, 43]. Auf Basis dieser Tierstudien wurde der humanisierte IgG1-mAK Prasinezumab entwickelt, der sich gegen das C-terminale Ende von aSyn richtet und lösliches und nichtlösliches aggregiertes aSyn bindet. In einer Phase-I-Studie an Gesunden war der Antikörper nach einmaliger intravenöser Applikation von 0,3 mg/kg bis 30,0 mg/kg sicher und gut verträglich. Die durchschnittliche maximale Konzentration im Serum lag zwischen 7,6 µg/ml bei einer Dosis von 0,3 mg/kg und 578,0 µg/ml bei einer Dosis von 30 mg/kg. Die durchschnittliche terminale Plasmahalbwertszeit betrug 18,2 Tage. Das ungebundene aSyn im Serum wurde bereits eine Stunde nach Ende der Antikörper-Infusion dosisabhängig um bis zu 96,5 % reduziert [59]. Die dreimalige Gabe des Antikörpers (Einzeldosis 0,3 mg/kg bis 60,0 mg/kg) bei 80 PD-Patienten zeigte ebenfalls eine gute Sicherheit und Verträglichkeit. Die terminale Plasmahalbwertszeit betrug im Schnitt 10,2 Tage. Ähnlich der vorangehenden Studie wurde das ungebundene aSyn im Serum um bis zu 97 % reduziert. Die Antikörper-Konzentration im Liquor entsprach im Mittel 0,3 % der Serumkonzentration, eine Reduktion von aSyn im Liquor konnte nicht gezeigt werden [29].
Die Phase-II-Studie PASADENA untersuchte als primären Endpunkt den Unterschied in der Summe der „Movement Disorder Society-Unified Parkinson’s Disease Rating Scale“ (MDS-UPDRS) Teile I–III im Vergleich zu Studienbeginn. 316 Patienten mit Morbus Parkinson erhielten alle vier Wochen intravenös Prasinezumab oder Placebo über 52 Wochen bis zu fünf Jahren. Dabei wurde nach bisherigen Ergebnissen der primäre Endpunkt verfehlt, es zeigte sich aber eine leichte Verzögerung der motorischen Verschlechterung [51, 52].
Obwohl die Ergebnisse der PASADENA-Studie keine zulassungsrelevanten Daten liefern konnten, waren sie Ausgangspunkt für eine weitere Studie mit Prasinezumab, der PADOVA-Studie (NCT04777331). Bei dieser Phase-IIb-Studie erhielten Patienten mit noch frühem, aber doch im Vergleich zu PASADENA weiter fortgeschrittenem Morbus Parkinson alle vier Wochen intravenös Prasinezumab oder Placebo. Primärer Endpunkt war die Zeit bis zur Entwicklung einer klinisch relevanten Verschlechterung, die als Zunahme von mindestens fünf Punkten im MDS-UPDRS Teil III definiert wurde. Die Finalisierung der Studie wird gegenwärtig im Jahr 2024 angestrebt (NCT04777331).
Der humane IgG1-mAK Cinpanemab (BIIB054) bindet N-terminal und hat eine deutlich höhere Affinität für aggregiertes als für monomeres aSyn [74]. In einer Phase-I-Studie erhielten gesunde Probanden und PD-Patienten einmalig 1 mg/kg bis 135 mg/kg des Antikörper. Bei einem Probanden zeigte sich in der im Rahmen der Studie durchgeführten Magnetresonanztomographie vier Wochen nach Infusion eine klinisch stumme zerebrale Ischämie, ansonsten kam es zu keinen gravierenden Nebenwirkungen. Die terminale Plasmahalbwertszeit lag zwischen 27,7 und 34,8 Tagen, die Antikörper-Konzentration im Liquor betrug 0,13 % bis 0,56 % der Serumkonzentration [7]. Die Phase-II-Studie SPARK wurde 2021 beendet, verfehlte jedoch den primären Endpunkt (Unterschied der Summe des MDS-UPDRS Teile I–III 52 und 72 Wochen nach Studienbeginn) im Vergleich zur Placebo-Gruppe. Es zeigte sich auch kein wesentlicher Einfluss der Antikörper-Therapie auf die sekundären Endpunkte (MDS-UPDRS-Subscores), sodass die Entwicklung von Cinpanemab beendet wurde (NCT03318523).
Im Gegensatz zum N-terminal bindenden Cinpanemab, ist MEDI1341 ein C-terminal bindender IgG1-mAK, der im Mausmodell die Akkumulation und Propagation von aSyn in Axonen reduzierte [63]. Eine Phase-I-Studie mit gesunden Probanden wurde 2021 abgeschlossen, bisher liegen hier keine Ergebnisse vor, eine weitere Phase-I-Studie mit PD-Patienten rekrutiert gegenwärtig noch nicht (NCT04449484).
ABBV-0805 ist ein humanisierter IgG4-mAK, der ebenfalls eine hohe Affinität zu löslichem aggregiertem aSyn aufweist und in einem Mausmodell zur Reduktion von aSyn-Aggregation führte. Eine Phase-I-Studie wurde aber vom Sponsor aus strategischen Überlegungen zurückgezogen [48] (NCT04127695). Ein weiterer humanisierter mAK gegen IgG1 ist Lu AF82 422, der monomeres und aggregiertes aSyn bindet und in Tiermodellen die Ausbreitung der aSyn-Pathologie reduzierte [20]. Eine Phase-I-Studie bei gesunden Probanden und PD-Patienten ist abgeschlossen, die Ergebnisse sind noch ausstehend, eine Phase-II-Studie bei Patienten mit MSA rekrutiert gegenwärtig (NCT03611569, NCT05104476).
Ausblick
Die Zahl der aktuell untersuchten Immuntherapien zeigt, wie dynamisch sich dieses Feld im Moment entwickelt und welches Potenzial diesen Therapieverfahren zugesprochen wird. Auch wenn die bisher durchgeführten Studien in den meisten Fällen eine gute Sicherheit der Antikörper-basierten Immuntherapien zeigen konnten, bleiben viele Fragen offen. Vor allem die Frage nach dem Zusammenhang zwischen peripheren aSyn-Spiegeln und einer möglichen Krankheitsprogression ist nach wie vor nicht gelöst. Zwar bauen die meisten Antikörper-Therapien im Blut ausreichende Konzentrationen auf, um eine Senkung der aSyn-Spiegel zu erreichen, im Liquor stellen sich diese Konzentrationen jedoch viel niedriger dar. Ob es allein ausreicht, durch ein Konzentrationsgefälle vom Liquor zum Blut auch zentral die Konzentration von aSyn zu senken, ist nicht ausreichend geklärt. Außerdem ist nicht in allen Fällen eindeutig, ob und welche Wirkungen die Antikörper intrazellulär haben könnten. Und letztlich ist trotz der überwältigenden Evidenz für eine pathogene Funktion des aggregierten aSyn nach wie vor nicht erwiesen, dass die Reduktion der aSyn-Konzentration im ZNS tatsächlich auch die Krankheitsprogression bei PD-Patienten beeinflusst. Dies muss vor allem vor dem Hintergrund gesehen werden, dass jedwede Immuntherapie bei idiopathischen Fällen erst sehr spät im Verlauf der Ausbreitung der aSyn-Pathologie zum Einsatz kommt. Zum Zeitpunkt der ersten motorischen Symptome befinden sich PD-Patienten bereits im Braak-Stadium III, haben bereits etwa 50 % ihrer dopaminergen striatalen Terminalen und etwa 30 % der dopaminergen Neurone in der Substantia nigra verloren [8]. Somit ist nicht klar, ob eine solche Immuntherapie nicht eigentlich zu spät kommt, um eine klinisch relevante Wirkung zu erreichen. Studien mit präsymptomatischen Genmutationsträgern könnten hier mehr Klarheit in das Wirkprinzip bringen, allerdings fehlt zurzeit ein Biomarker, der den Zeitpunkt des Therapiebeginns eingrenzen könnte. Solche präsymptomatischen Behandlungsregimes werden gegenwärtig bei einer anderen neurodegenerativen Erkrankung, der amyotrophen Lateralsklerose (ALS), untersucht (NCT04856982) und könnten auch für Parkinson-Patienten wertvolle Informationen für künftige Studiendesigns liefern.
Abkürzungsverzeichnis
AK: Antikörper
AD: Alzheimer-Erkrankung (engl. Alzheimer’s disease)
aSyn: Alpha-Synuclein
BHS: Blut-Hirn-Schranke
EMA: European Medicines Agency
FDA: Food and Drug Administration
mAK: monoklonaler Antikörper
MDS-UPDRS: Movement Disorder Society-Unified Parkinson’s Disease Rating Scale
MSA: Multisystematrophie
PD: Morbus Parkinson (engl. Parkinson’s disease)
Interessenkonflikterklärung
PL: Honorare für die Beratung oder Teilnahme an einem Expertenbeirat von AbbVie, ITF Pharma, Stadapharm, Woolsey Pharmaceuticals; Autoren- oder Vortragshonorare von Alexion, Bial, Desitin; Drittmittel von BMBF, DFG, DGM, DZNE, StMWK; Patente EP 2825175 B1 und US 9980972 B2
GM, AW: Keine Interessenkonflikte
Literatur
1. Bayer AJ, Bullock R, Jones RW, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67.
2. Bergström AL, Kallunki P, Fog K. Development of passive immunotherapies for synucleinopathies. Mov Disord 2016;31:203–13. doi: 10.1002/mds.26481.
2a. Billingley KJ, et al. Genetic risk factors in Parkinson’s disease. Cell Tissue Res 2018;373:9–20. doi: 10.1007/s00441-018-2817-y.
3. Birol M, Wojcik SP, Miranker AD, Rhoades E. Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated α-Synuclein. bioRxiv Published online 2018: 1–29. https://doi.org/10.1101/407247.
4. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol 2020;19:170–8. doi: 10.1016/S1474-4422(19)30287-X.
5. Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet 2021;397:2284–2303. doi: 10.1016/S0140-6736(21)00218-X.
6. Braak H, Tredici K del, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9.
7. Brys M, Fanning L, Hung S, et al. Randomized phase I clinical trial of anti-α-synuclein antibody BIIB054. Mov Disord 2019;34:1154–63. doi: 10.1002/mds.27738.
8. Burke RE, O’Malley K. Axon degeneration in Parkinson’s disease. Exp Neurol 2013;246:72–83. doi: 10.1016/j.expneurol.2012.01.011.
9. Chartier-Harlin MC, Kachergus J, Roumier C, et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1.
10. Chatterjee D, Kordower JH. Immunotherapy in Parkinson’s disease: current status and future directions. Neurobiol Dis 2019;132. doi: 10.1016/j.nbd.2019.104587.
11. Chen Z, Yang Y, Yang X, et al. Immune effects of optimized DNA vaccine and protective effects in a MPTP model of Parkinson’s disease. Neurol Sci 2013;34:1559–70. doi: 10.1007/s10072-012-1284-6.
12. Dauer Née Joppe K, Tatenhorst L, Caldi Gomes L, et al. Brain iron enrichment attenuates α-synuclein spreading after injection of preformed fibrils. J Neurochem 2021;159:554–73. doi: 10.1111/jnc.15461.
13. Davtyan H, Zagorski K, Petrushina I, et al. MultiTEP platform-based DNA vaccines for alpha-synucleinopathies: preclinical evaluation of immunogenicity and therapeutic potency. Neurobiol Aging 2017;59:156–70. doi: 10.1016/j.neurobiolaging.2017.08.006.
14. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A antibody alters CNS and plasma A clearance and decreases brain A burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 2001;98:8850–5. doi: 10.1073/pnas.151261398.
15. Deuschl G, Beghi E, Fazekas F, et al. The burden of neurological diseases in Europe: an analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020;5:e551–e567. doi: 10.1016/S2468-2667(20)30190-0.
16. Dickson DW. Neuropathology of Parkinson disease. Parkinsonism Relat Disord 2018;46:S30–S33. https://doi.org/10.1016/j.parkreldis.2017.07.033.
17. Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 2002;5:452–7. doi: 10.1038/nn842.
18. Doucet M, El-Turabi A, Zabel F, et al. Preclinical development of a vaccine against oligomeric alpha-synuclein based on virus-like particles. PLoS One 2017;12:e0181844. doi: 10.1371/journal.pone.0181844.
19. Espay AJ, Vizcarra JA, Marsili L, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019;92:329–37. doi: 10.1212/WNL.0000000000006926.
20. Fjord-Larsen L, Thougaard A, Wegener KM, et al. Nonclinical safety evaluation, pharmacokinetics, and target engagement of Lu AF82422, a monoclonal IgG1 antibody against alpha-synuclein in development for treatment of synucleinopathies. MAbs 2021;13. doi: 10.1080/19420862.2021.1994690.
21. Games D, Valera E, Spencer B, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 2014;34:9441–54. doi: 10.1523/JNEUROSCI.5314-13.2014.
22. Ghochikyan A, Petrushina I, Davtyan H, et al. Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-synuclein: Feasibility study. Neurosci Lett 2014;560:86–91. doi: 10.1016/j.neulet.2013.12.028.
23. Grozdanov V, Danzer KM. Release and uptake of pathologic alpha-synuclein. Cell Tissue Res 2018;373:175–82. doi: 10.1007/s00441-017-2775-9.
24. Grudina C, Kouroupi G, Nonaka T, Hasegawa M, Matsas R, Zurzolo C. Human NPCs can degrade α–syn fibrils and transfer them preferentially in a cell contact-dependent manner possibly through TNT-like structures. Neurobiol Dis 2019;132:104609. doi: 10.1016/j.nbd.2019.104609.
25. Gustavsson A, Svensson M, Jacobi F, et al. Cost of disorders of the brain in Europe 2010. Eur Neuropsychopharmaco 2011;21:718–79. doi: 10.1016/j.euroneuro.2011.08.008.
26. Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener 2020;15:1–12. doi: 10.1186/s13024-020-00368-6.
27. Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 2013;110. doi: 10.1073/pnas.1301440110.
28. Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. Bivalent brain shuttle increases antibody uptake by monovalent binding to the transferrin receptor. Theranostics 2017;7:308–18. doi: 10.7150/thno.17155.
29. Jankovic J, Goodman I, Safirstein B, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol 2018;75:1206–14. doi: 10.1001/jamaneurol.2018.1487.
30. Karagiannidis C, Lang K, Mikolajewska A, Malin JJ, Kluge S, Spinner CD. Therapie und Prophylaxe: Antikörper gegen COVID-19. Dtsch Arztebl 2021;118:2212.
31. Kim C, Hovakimyan A, Zagorski K, et al. Efficacy and immunogenicity of MultiTEP-based DNA vaccines targeting human α-synuclein: prelude for IND enabling studies. NPJ Vaccines 2022;7:1. doi: 10.1038/s41541-021-00424-2.
32. Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 2021;17:696–701. https://doi.org/10.1002/alz.12213.
33. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 2008;14:504–6. doi: 10.1038/nm1747.
34. Lee JS, Lee SJ. Mechanism of anti-α-synuclein immunotherapy. J Mov Disord 2016;9:14–9. doi: 10.14802/jmd.15059.
35. Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 2008;14:501–3. doi: 10.1038/nm1746.
36. Lindestam Arlehamn CS, Dhanwani R, Pham J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 2020;11:1875. doi: 10.1038/s41467-020-15626-w.
37. Malfertheiner K, Stefanova N, Heras-Garvin A. The concept of α-synuclein strains and how different conformations may explain distinct neurodegenerative disorders. Front. Neurol 2021;12:1–10. doi: 10.3389/fneur.2021.737195.
38. Mandler M, Rockenstein E, Overk C, et al. Effects of single and combined immunotherapy approach targeting amyloid β protein and α-synuclein in a dementia with Lewy bodies-like model. Alzheimers Dement 2019;15:1133–48. doi: 10.1016/j.jalz.2019.02.002.
39. Mandler M, Valera E, Rockenstein E, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener 2015;10:10. doi: 10.1186/s13024-015-0008-9.
40. Mandler M, Valera E, Rockenstein E, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol 2014;127:861–79. doi: 10.1007/s00401-014-1256-4.
41. Mao X, Ou MT, Karuppagounder SS, et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016;353. doi: 10.1126/science.aah3374.
42. Masliah E, Rockenstein E, Adame A, et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005;46:857–68. doi: 10.1016/j.neuron.2005.05.010.
43. Masliah E, Rockenstein E, Mante M, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 2011;6:e19338. doi: 10.1371/journal.pone.0019338.
44. McClendon S, Rospigliosi CC, Eliezer D. Charge neutralization and collapse of the C-terminal tail of alpha-synuclein at low pH. Protein Sci 2009;18:1531–40. doi: 10.1002/pro.149.
45. Meissner WG, Traon AP, Foubert‐Samier A, et al. A phase 1 randomized trial of specific active α‐synuclein immunotherapies PD01A and PD03A in multiple system atrophy. Mov Disord 2020;35:1957–65. doi: 10.1002/mds.28218.
46. Nimmo JT, Smith H, Wang CY, et al. Immunisation with UB-312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric α-synuclein accumulation in the brain and gut. Acta Neuropathol 2022;143:55–73. doi: 10.1007/s00401-021-02381-5.
47. Nimmo JT, Verma A, Dodart JC, et al. Novel antibodies detect additional α-synuclein pathology in synucleinopathies: potential development for immunotherapy. Alzheimers Res Ther 2020;12:159. doi: 10.1186/s13195-020-00727-x.
48. Nordström E, Eriksson F, Sigvardson J, et al. ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents build-up of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol Dis. 2021;161. https://doi.org/10.1016/j.nbd.2021.105543.
49. Oliveira LMA, Gasser T, Edwards R, et al. Alpha-synuclein research: defining strategic moves in the battle against Parkinson’s disease. NPJ Parkinsons Disease 2021;7:65. doi: 10.1038/s41531-021-00203-9.
50. Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003;61:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8.
51. Pagano G, Boess FG, Taylor KI, et al. A phase II study to evaluate the safety and efficacy of prasinezumab in early Parkinson’s disease (PASADENA): rationale, design, and baseline data. Front Neurol 2021;12:1–17. doi: 10.3389/fneur.2021.705407.
52. Pagano G, Taylor K, Cabrera J, et al. PASADENA: A phase 2 study to evaluate the safety and efficacy of prasinezumab in early Parkinson’s disease; Part 1 Week-52 results. MDS Virtual Congress 2020; Abstract Number: 928.
53. Poewe W, Volc D, Seppi K, et al. Safety and tolerability of active immunotherapy targeting α-synuclein with PD03A in patients with early Parkinson’s disease: a randomized, placebo-controlled, phase 1 study. J Parkinsons Dis 2021;11:1079–89. doi: 10.3233/JPD-212594.
54. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–47. doi: 10.1126/science.276.5321.2045.
55. Rabinovici GD. Controversy and progress in Alzheimer’s disease – FDA approval of aducanumab. N Engl J Med 2021;385:771–4. doi: 10.1056/NEJMp2111320.
56. Reyes JF, Sackmann C, Hoffmann A, et al. Binding of α-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol 2019;138:23–47. doi: 10.1007/s00401-019-02007-x.
57. Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 2022;79:13. doi: 10.1001/jamaneurol.2021.4161.
58. Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci 2002;3:824–8. doi: 10.1038/nrn938.
59. Schenk DB, Koller M, Ness DK, et al. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 2017;32:211–8. doi: 10.1002/mds.26878.
60. Schneeberger A, Mandler M, Mattner F, Schmidt W. AFFITOME® technology in neurodegenerative diseases: the doubling advantage. Hum Vaccin 2010;6:948–52. doi: 10.4161/hv.6.11.13217.
61. Schneeberger A, Tierney L, Mandler M. Active immunization therapies for Parkinson’s disease and multiple system atrophy. Mov Disord 2016;31:214–24. doi: 10.1002/mds.26377.
62. Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 2017;32:1504–23. doi: 10.1002/mds.27193.
63. Schofield DJ, Irving L, Calo L, et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol Dis 2019;132:104582. doi: 10.1016/j.nbd.2019.104582.
64. Scudamore O, Ciossek T. Increased oxidative stress exacerbates α-synuclein aggregation in vivo. J Neuropathol Exp Neurol 2018;77:443–53. doi: 10.1093/jnen/nly024.
65. Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature Genetics 2009;41:1308–12. doi: 10.1038/ng.487.
66. Singleton AB, Farrer M, Johnson J, et al. α-synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841. doi: 10.1126/science.1090278.
67. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–40. doi: 10.1038/42166.
68. Stoessl AJ. Immunotherapy for Parkinson’s disease: stay tuned. Lancet Neurol 2020;19:561–2. doi: 10.1016/S1474-4422(20)30175-7.
69. Sulzer D, Edwards RH. The physiological role of α-synuclein and its relationship to Parkinson’s disease. J Neurochem 2019;150:475–86. doi: 10.1111/jnc.14810.
70. Suzuki M, Sango K, Wada K, Nagai Y. Pathological role of lipid interaction with α-synuclein in Parkinson’s disease. Neurochem Int 2018;119:97–106. doi: 10.1016/j.neuint.2017.12.014.
71. Ugen KE, Lin X, Bai G, et al. Evaluation of an α synuclein sensitized dendritic cell based vaccine in a transgenic mouse model of Parkinson disease. Hum Vaccin Immunother 2015;11:922–30. doi: 10.1080/21645515.2015.1012033.
72. Vicario M, Cieri D, Brini M, Calì T. The close encounter between alpha-synuclein and mitochondria. Front Neurosci 2018;12. doi: 10.3389/fnins.2018.00388.
73. Volc D, Poewe W, Kutzelnigg A, et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol 2020;19:591–600. doi: 10.1016/S1474-4422(20)30136-8.
74. Weihofen A, Liu YT, Arndt JW, et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol Dis 2019;124:276–88. doi: 10.1016/j.nbd.2018.10.016.
75. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 2017;23:1–13. doi: 10.1038/nm.4269.
* Diese Autoren haben gleichermaßen beigetragen.
Prof. Dr. Paul Lingor, Technische Universität München, Fakultät für Medizin, Klinikum rechts der Isar, Klinik und Poliklinik für Neurologie, Ismaninger Str. 22, 81675 München; DZNE, Deutsches Zentrum für Neurodegenerative Erkrankungen, Standort München; SyNergy, Cluster für Systemneurologie, München, E-Mail: paul.lingor@tum.de
Dr. med. univ. Gerrit Machetanz, Dr. med. Andreas Wolff, Technische Universität München, Fakultät für Medizin, Klinikum rechts der Isar, Klinik und Poliklinik für Neurologie
Antibody-based immunotherapies for Parkinson’s disease
Currently, there are no disease-modifying therapies available for patients suffering from Parkinson’s disease (PD) and inevitably their motor and non-motor symptoms are progressing over time. Aggregation of the misfolded protein alpha-synuclein (aSyn) is considered a hallmark in the pathogenesis of PD. Aggregated aSyn can propagate from cell-to-cell in the peripheral and central nervous system. For almost twenty years research has been conducted into immunotherapies primarily targeting aggregated aSyn and its propagation. Several approaches using active or passive immunization are now being investigated in clinical trials. In this review, we are discussing the molecular background, the current state, and challenges in the development of antibody-based immunotherapies for PD.
Key words: Parkinson’s disease, antibody, alpha-synuclein, immunotherapy, neurodegeneration
Psychopharmakotherapie 2022; 29(02):56-63