Michael Paulzen*, Claus Liebe*, Aachen, Christoph Hiemke, Mainz, und Georgios Schoretsanitis, Zürich/New York
Pharmakoepidemiologische Daten belegen zunehmende Verordnungsraten für Depot-Antipsychotika (long-acting injectable antipsychotics, LAI) hauptsächlich der sogenannten zweiten Generation [33, 46, 60a], sowohl bei der Behandlung von Patienten mit Erkrankungen aus dem schizophrenen Formenkreis als auch bei Patienten mit bipolaren Störungen [54]. Mittlerweile sind Depot-Formulierungen sowohl für Antipsychotika der ersten (first generation antipsychotics, FGA) als auch der zweiten (second generation antipsychotics, SGA) Generation verfügbar.
Insbesondere bei der Behandlung spezifischer Patientenkollektive (z. B. therapierefraktäre schizophrene Spektrumserkrankungen, Patienten mit Adhärenzproblemen, bestehende Komorbiditäten einschließlich Suchterkrankungen) oder bei aggressivem oder suizidalem Verhalten können Depot-Antipsychotika ein wertvoller Bestandteil der Therapie sein [2, 22, 38, 77]. Ferner zeigen verschiedene Beobachtungsstudien einen klaren Vorteil von Depot-Formulierungen im Vergleich zu oralen Antipsychotika in Bezug auf geringere Rehospitalisierungsraten sowie ein deutlich niedrigeres Rezidivrisiko [50, 55]. Auch bei Erstmanifestationen schizophrener Störungen (first-episode schizophrenia) können Depot-Antipsychotika eingesetzt werden, um einer möglichen Adhärenz-Problematik vorzubeugen [78]. Für die Kombination von zwei Depot-Antipsychotika ist positive klinische Erfahrung zumindest aus Fallberichten zu entnehmen [103].
Für eine erfolgreiche Umstellung auf ein Depot-Antipsychotikum ist zunächst das Ansprechen auf die orale Darreichungsform eine wesentliche Voraussetzung [60]. Nach der ersten Applikation der Depot-Injektion steigt die Wirkstoffkonzentration in aller Regel nur langsam an (Abb. 1). Daher ist in der Umstellungsphase meist noch eine passagere orale Gabe erforderlich, um einen drastischen Abfall der Wirkstoffkonzentration zu vermeiden; allerdings wird dieser Aspekt in der klinischen Routine oft nicht ausreichend beachtet [5]. Ein zielgerichteter Einsatz von TDM bei der Umstellung von einem oralen Antipsychotikum auf ein Depot-Präparat kann erschwert werden, wenn die Zeit zwischen Probeneingang und Befunderstellung seitens der Labore verlängert ist. Mittlerweile sind die meisten etablierten TDM-Labore jedoch in der Lage, mehrfach wöchentlich die Wirkstoffkonzentrationen gängiger Antipsychotika zu bestimmen.
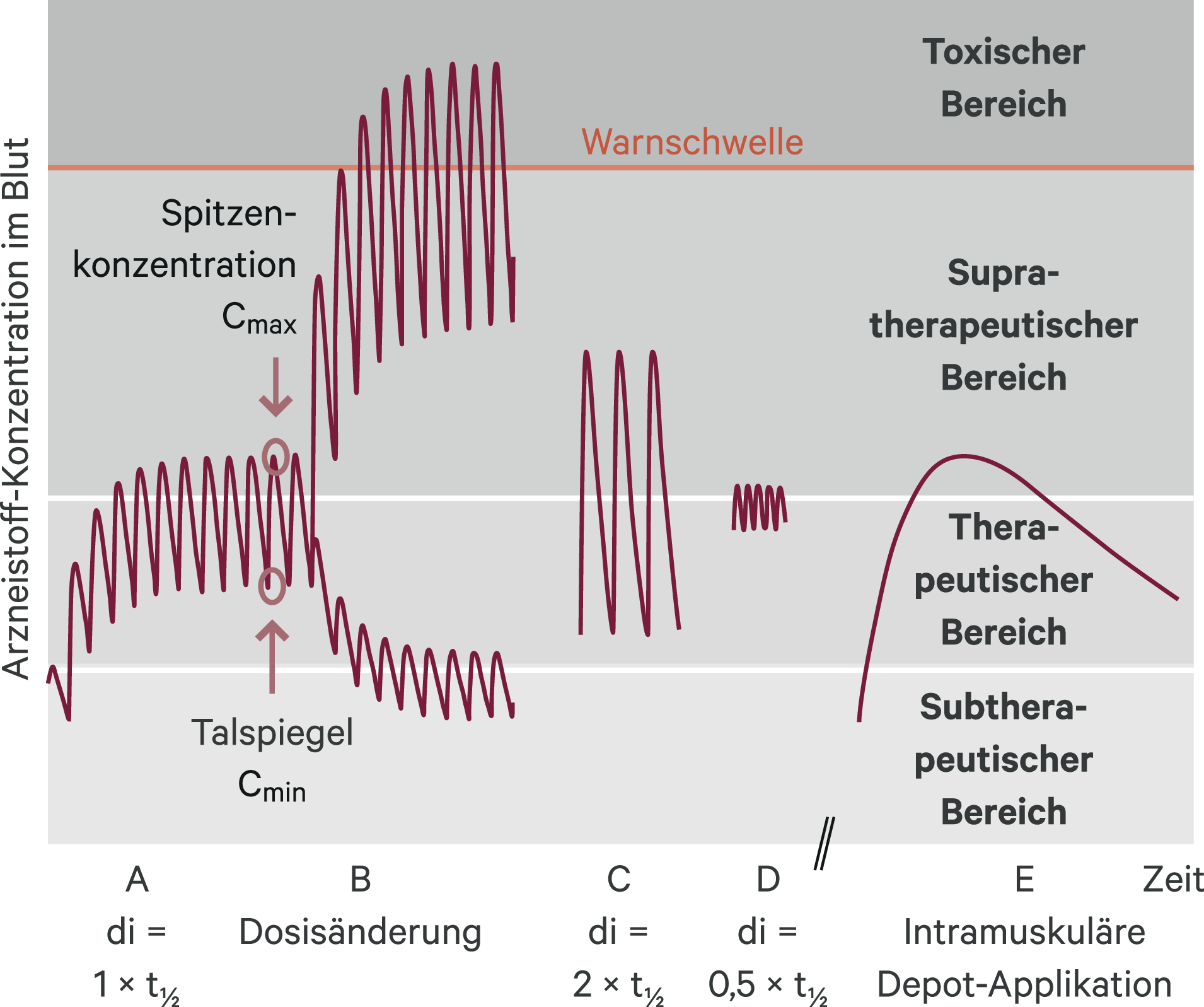
Abb. 1. Konzentration-Zeit-Kurve eines Arzneistoffs nach oraler Gabe oder intramuskulärer Depot-Applikation. Die Abbildung zeigt einen hypothetischen Arzneistoff mit einem gleichen Dosisintervall (di) zu dessen Halbwertszeit (di = t1/2); eine Situation, die für viele Arzneistoffe betrachtet wird (z. B. t1/2 = 12 h, di = 12 h, Kurve A). Intramuskuläre Depot-Applikation: Spitzen-Konzentrationen des Arzneistoffs können in Abhängigkeit von der Formulierung frühestens nach einem Tag und spätestens nach vier Wochen erreicht werden. Talspiegel-Konzentrationen sind kurz vor der nächsten Applikation gegeben. Die Blutentnahme während der Eliminationsphase nach vollständiger Absorption (Maximum) resultiert trotz gleicher Fläche unter der Kurve („Area under the Curve“ [AUC]) in höheren Werten, im Vergleich zu der Messung der Talspiegel nach oraler Applikation [34].
Leitlinien zu Äquivalenzdosierungen für die Umstellung von oralen auf Depot-Antipsychotika existieren beispielsweise für Antipsychotika der zweiten Generation (SGA) [26, 39, 60]. Diese basieren meist auf pharmakokinetischen Modellen oder simulierten Daten pharmazeutischer Hersteller. Ergänzt und erweitert werden die Umstellempfehlungen durch zunehmende klinische Daten [20, 30, 83]. Diese bilden die Grundlage für zukünftige Orientierungshilfen zur weiteren Verbesserung der Therapiesicherheit von Depot-Antipsychotika [86, 87, 95, 96]. Für Antipsychotika der ersten Generation (FGA) liegen TDM-basierte Übergangsstudien von oralen Applikationsformen zu Depot-Antipsychotika vor, obwohl diese im Laufe der Jahre immer seltener verordnet und angewandt wurden [71].
Die vorliegende Arbeit fasst das aktuelle Wissen über TDM bei der Anwendung von Depot-Antipsychotika zusammen [84] und schafft einen Rahmen zur praktischen Anwendung von TDM bei der Umstellung von oralen auf Depot-Antipsychotika.
Physikochemische Eigenschaften von Depot-Antipsychotika
Depot-Antipsychotika unterscheiden sich untereinander zum Teil deutlich in ihrer chemischen Struktur. Jedoch liegen die meisten Depot-Antipsychotika in öliger Lösung als Ester in Verbindung mit einer Fettsäure (z. B. Decansäure) vor [56]. Beispiele hierfür sind die Decanoate von Haloperidol, Flupentixol, Fluphenazin, Perphenazin und Zuclopenthixol, sowie Perphenazinenantat, Zuclopenthixolacetat und Paliperidonpalmitat. Alle veresterten Depot-Antipsychotika sind somit Prodrugs. Aufgrund der raschen Hydrolyse veresterter Verbindungen sind die Konzentrationen der Ausgangssubstanzen im Plasma nicht bestimmbar. Die hier beschriebenen Wirkstoffkonzentrationsbestimmungen beziehen sich auf die jeweilige Substanz nach erfolgter Hydrolyse.
Durch Veresterung und Verbindung mit langkettigen Fettsäuren wird einerseits die Wasserlöslichkeit verringert und andererseits der Verteilungskoeffizient und die Löslichkeit in Ölen erhöht. Als Öl wird vor allem Sesamöl wegen seiner hohen Viskosität und guten Verträglichkeit verwendet [76]. Nach der Injektion in das Muskelgewebe wird der Wirkstoff langsam ins Blut freigesetzt [58].
Neuere Formulierungen wie Aripiprazol-Monohydrat (AM) oder Risperidon-ISM® erzielen eine Retardierung durch die Bildung eines festen polymeren Matrixsystems, welches die Auflösungs- und Diffusionsgeschwindigkeit nach Injektion reguliert.
Zweitgenerations-Depot-Antipsychotika liegen meist als Formulierungen aus wenig wasserlöslichen Kristallen der Ausgangsverbindungen oder der Prodrugs vor. Die Freisetzung der Wirkstoffe wird dabei in erster Linie durch die geringe Wasserlöslichkeit des Wirkstoffs einerseits und die langsame Auflösung der Kristallpartikel andererseits beeinflusst. Die Löslichkeit steigt dabei mit zunehmender Oberflächengröße, welche durch die abnehmende Partikelgröße erzielt wird [82]. Durch ein Gleichgewicht zwischen Partikelgröße und Löslichkeit lässt sich die Pharmakokinetik einer Substanz gezielt beeinflussen, wodurch es über längere Zeiträume möglich wird, Wirkstoffkonzentrationen in einem gewünschten Bereich, dem therapeutischen Referenzbereich, aufrechtzuerhalten. Die gezielten Veränderungen galenischer Eigenschaften ist eine klare Innovation im Bereich der Depot-Antipsychotika.
Ein detaillierter Überblick über Dosierungsbereiche, Halbwertszeiten, die Notwendigkeit einer oralen Supplementierung, zu berücksichtigende Merkmale und pharmakokinetische/pharmakodynamische Eigenschaften, Wechselwirkungen und dosisabhängige Wirkstoffkonzentrationen (C/D) für Depot-Antipsychotika der ersten Generation findet sich in Tabelle 1, für Depot-Antipsychotika der zweiten Generation entsprechend in Tabelle 2.
Antipsychotika der ersten Generation (FGA)
Tab. 1. Dosisbereiche, Halbwertszeiten, Zeitdauer bis Erreichen eines Steady-States, Notwendigkeiten oraler Überlappungen, relevante pharmakokinetische und pharmakodynamische Interaktionen, dosisbezogene Konzentrationen (C/D) oraler Darreichungsformen und therapeutische Referenzbereiche (TRR) für Depot-Antipsychotika der ersten Generation (nach [84])
|
Dosis [mg] |
t1/2 [Tage] |
Steady-State |
Orale Überlappung |
Dosisanpassung |
Potenzielle Interaktionen (PK oder PD) |
C/D |
TRR [ng/ml] |
|
|
Flupentixoldecanoat |
4-fache orale Dosis alle 2 Wochen 8-fache orale Dosis alle 4 Wochen |
17 |
3 Monate |
7 Tage |
Alkohol, Antiarrhythmika, Barbiturate, Erythromycin, Guanethidin, Hypnotika, Levodopa, Metoclopramid, Moxifloxacin, Trizyklika, Thiaziddiuretika, Thioridazin |
0,32 |
0,5–5 |
|
|
Fluphenazindecanoat |
12,5–100/2 Wochen |
14 |
6 Wochen |
Ja |
Atropin |
0,07 |
1–10 |
|
|
Fluspirilen |
2–6/Woche (ambulant) 3–8/Woche (stationär) |
7–14 |
Unklar |
Nein |
Alter |
Alkohol, Hypnotika, Antihistaminika, Analgetika, Polymyxin B, Benzatropin, Trihexyphenidyl, Phenobarbital, Carbamazepin, Diphenylhydantoin, Lithium, Levodopa, Metoclopramid, Epinephrin, Atropin, Disulfiram, Pentetrazol, Gonadorelin |
k. A. |
0,1–2,2 |
|
Haloperidoldecanoat |
25–300/alle 4 Wochen |
17 |
3 Monate |
Ja |
CYP2D6 PM Alter |
CYP2D6-Inhibitoren (Fluoxetin, Paroxetin) CYP3A4-Induktoren (Carbamazepin) Lithium, Antikonvulsiva, Antikoagulanzien, Ketoconazol, Itraconazol, Nefazodon, Buspiron, Venlafaxin, Alprazolam, Fluvoxamin, Chinidin, Sertralin, Chlorpromazin, Promethazin, Rifampicin, Clozapin |
0,81 |
1–10 |
|
Perphenazindecanoat |
50–200/alle 2 Wochen |
14 |
3 Monate |
Nicht erforderlich |
CYP2D6-Inhibitoren (Fluoxetin, Paroxetin) |
0,05 |
0,6–2,4 |
|
|
Perphenazinenanthat |
25–300/alle 2 Wochen |
4–6 |
1 Monat |
Nicht erforderlich |
CYP2D6-Inhibitoren (Fluoxetin, Paroxetin) |
0,05 |
0,6–2,4 |
|
|
Zuclopenthixolacetat |
50–150 |
1–2 |
Unklar |
Unklar |
CYP2D6-Inhibitoren Alkohol, Barbiturate, Guanethidin, Metoclopramid, Levodopa, Trizyklika |
0,42 |
4–50 |
|
|
Zuclopenthixol decanoat |
50–400/alle 2 Wochen |
7–21 |
2 Monate |
Ja |
CYP2D6-Inhibitoren Alkohol, Barbiturate, Guanethidin, Metoclopramid, Levodopa, Trizyklika |
0,42 |
4–50 |
CYP: Cytochrom P450; C/D: dosisbezogene Konzentrationen oraler Darreichungsformen; PD: Pharmakodynamik; P-gp: P-Glykoprotein; PK: Pharmakokinetik; PM: Poor Metabolizer; W: Woche(n)
Flupentixoldecanoat
Flupentixol selbst ist ein hochpotentes Antipsychotikum aus der Gruppe der Thioxanthene mit nur geringer sedierender Wirkung. Flupentixoldecanoat ist ein Prodrug, ein Decansäureester des Flupentixols. Die Veresterung mit Decansäure führt zu einer langsamen Freisetzung durch Diffusion aus der öligen Lösung sowie durch metabolischen Abbau des Öls im Injektionsbereich. Nach der Freisetzung aus dem Muskelgewebe wird die Substanz hydrolysiert [28]. Der Wirkeintritt erfolgt innerhalb von 24 bis 72 Stunden nach der Injektion, die höchsten Serumkonzentrationen werden zwischen drei und 17 Tagen nach der Injektion erreicht. Aufgrund der langsamen Freisetzung ist die Halbwertszeit nach einer einmaligen Verabreichung (im Vergleich zu oralem Flupentixol) auf drei bis acht Tage und nach Mehrfachgabe auf bis zu 17 Tage verlängert [48]; Steady-State-Wirkstoffkonzentrationen werden erst nach zwei bis drei Monaten erreicht. Studien zeigen große Schwankungen der Plasmakonzentration während der Erhaltungstherapie [93, 102]. Basierend auf der maximalen/minimalen Fluktuation der Wirkstoffkonzentration wird ein Injektionsintervall von zwei bis vier Wochen empfohlen [79, 90]. Dabei ist eine patientenindividuelle Bestimmung des optimalen Intervalls erforderlich. Dies kann ein langwieriger Prozess sein, insbesondere, wenn allein aufgrund klinischer Symptome und ohne Einbezug der Wirkstoffkonzentration vorgegangen wird.
Fluphenazindecanoat
Fluphenazin ist ein Phenothiazin-Derivat vom Piperazin-Typ und ein hochpotentes FGA. Die Depot-Formulierung liegt als Decanoatester in Sesamöl-Suspension vor. Nach intramuskulärer Injektion der öligen Lösung wird Fluphenazindecanoat langsam zum nichtveresterten Fluphenazin aufgespalten [4]. Der Wirkeintritt erfolgt innerhalb von 24 bis 96 Stunden, die Eliminationshalbwertszeit beträgt sieben bis zehn Tage und erhöht sich nach mehrmaliger Gabe auf bis zu 14 Tage.
Fluspirilen
Fluspirilen gehört zur Klasse der Diphenylbutylpiperidine mit guter Absorptionsrate für Fett- und Hirngewebe. Es unterscheidet sich von anderen FGA-Depot-Antipsychotika, da es nicht mit einer Fettsäure verestert wird und somit auch kein Prodrug ist [3]. Das Medikament wird intramuskulär als mikronisierte Partikel in wässriger Suspension verabreicht. Nach einer Einzeldosis von 1,5 mg werden maximale Plasmakonzentrationen nach 24 Stunden erreicht. Die Elimination verläuft nicht linear. Nach einer einzelnen intramuskulär verabreichten Dosis wird weniger als 50 % in sieben Tagen ausgeschieden, und ungefähr 70 % des Wirkstoffs und der Metaboliten werden innerhalb von 27 Tagen ausgeschieden.
Haloperidoldecanoat
Haloperidoldecanoat ist ein Decansäureester von Haloperidol und somit ein Depot-Antipsychotikum aus der Substanzgruppe der Butyrophenone. Nach der Injektion wird es langsam aus dem Muskelgewebe freigesetzt und zu freiem Haloperidol hydrolysiert. Die Wirkstoffkonzentration erreicht an Tag 7 (3–9 Tage) nach der intramuskulären Injektion ihr Maximum. Die Halbwertszeit beträgt etwa drei Wochen. Die Metabolisierung von Haloperidol erfolgt im Wesentlichen unter Beteiligung von Cytochrom P450 (CYP) 3A4 und 2D6 [73]. Der Hauptmetabolit, reduziertes Haloperidol (Hydroxyhaloperidol), ist ein Enantiomer mit asymmetrischem Zentrum. Beide Konfigurationen verfügen über eine wesentlich geringere Affinität zu Dopamin-D2-Rezeptoren als Haloperidol. Im Gehirn beeinflusst der Metabolit den Umsatz von Dopamin in ähnlichem Maße wie Haloperidol [42]. Durch eine im Gehirn von Ratten und Menschen nachgewiesene Aktivität von CYP3A4 kann außerdem durch Oxidation eine Umwandlung zurück in die Muttersubstanz erfolgen [100].
Perphenazindecanoat and -enanthat
Perphenazindecanoat verfügt über eine Piperazin-Ethanol-Seitenkette, die durch eine Konjugation mit Carbonsäuren verestert wird. Perphenazinenanthat wird nach intramuskulärer Injektion langsam aus der öligen Depotform freigesetzt und schnell zu Perphenazin hydrolysiert. Das freie Perphenazin wird grundsätzlich in gleicher Weise metabolisiert wie oral verabreichtes Perphenazin. Die systemische Verfügbarkeit von Perphenazinenanthat ist jedoch deutlich größer als die der oralen Applikation, da im Unterschied zur oralen Gabe von Perphenazin ein First-Pass-Effekt entfällt. Nach regelhaft zweiwöchentlicher intramuskulärer Gabe von Perphenazinenanthat [7] steigen die Perphenazin-Konzentrationen im Blut rasch an [58] und erreichen das Maximum nach zwei bis drei Tagen. Halbmaximale Konzentrationen finden sich etwa fünf bis acht Tage nach Injektion. Die Eliminationshalbwertszeit von Perphenazindecanoat beträgt zwei Wochen, diejenige von Perphenazinenanthat vier bis sechs Wochen (vgl. Tab. 1).
Zuclopenthixolacetat und -decanoat
Die Acetatformulierung ist ein klares, gelbliches Öl. Die endständige Alkoholgruppe von Zuclopenthixol ist mit Essigsäure verestert [10]. Die Decanoat-Formulierung liegt ebenfalls als ölige Lösung vor, die Alkolholgruppe ist hier mit Decansäure verestert. Nach intramuskulärer Applikation wird Zuclopenthixoldecanoat langsam aus dem Öldepot freigesetzt und hydrolysiert. Die pharmakokinetischen Parameter unterscheiden sich deutlich zwischen den zwei Formulierungen. Die maximale Plasmakonzentration für die Acetat-Formulierung wird nach 36 Stunden erreicht, die der Decanoat-Variante nach vier bis sieben Tagen [1, 7, 91, 98].
Zuclopenthixolacetat hat eine Wirkdauer von zwei bis drei Tagen [10, 47], während Zuclopenthixoldecanoat zwei bis vier Wochen wirkt [7, 17, 47]. Basierend auf den Schwankungen der Plasmakonzentration von oral eingenommenem Zuclopenthixol wird ein Dosierungsintervall von drei Wochen für die Decanoat-Formulierung empfohlen [89].
Antipsychotika der zweiten Generation (SGA)
Tab. 2. Dosisbereiche, Halbwertszeiten, Zeitdauer bis Erreichen eines Steady-States, Notwendigkeiten oraler Überlappungen, relevante pharmakokinetische und pharmakodynamische Interaktionen, dosisbezogene Konzentrationen (C/D) oraler Darreichungsformen und therapeutische Referenzbereiche (TRR) für Depot-Antipsychotika der zweiten Generation (nach [84])
|
Dosis [mg] |
t1/2 [Tage] |
Steady-State |
Orale Überlappung |
Dosisanpassung |
Potenzielle Interaktionen (PK oder PD) |
C/D |
TRR [ng/ml] |
|
|
Aripiprazol-Lauroxil (AL) |
441–882/alle 2 W 882/alle 6 W 1064/alle 8 W |
53,9–57,2 |
Unklar |
21 Tage |
CYP2D6-PM oder -UM |
CYP3A4-Inhibitoren oder -Induktoren |
11,721 |
100–3501 |
|
Aripiprazolmonohydrat (AM) |
300–400/alle 4 W |
29,9–46,5 |
> 12 W |
14 Tage |
CYP2D6-PM oder -UM |
CYP3A4-Inhibitoren oder -Induktoren |
11,721 |
100–3501 |
|
Olanzapinpamoat |
150–210/alle 2 W 300–405/alle 4 W |
30 |
7 Monate |
Nein |
Geschlecht Rauchen Alter |
CYP1A2-Inhibitoren (z. B. Fluvoxamin) Alkohol, Antihypertensiva, Levodopa, Diazepam |
1,85 |
10–40 |
|
Paliperidonpalmitat (PP1M) |
25–150/alle 4 W |
25–49 |
8 Monate |
Nein |
Alter, Nierenfunktionsstörung |
CYP3A4- und P-gp-Induktoren (z. B. Carbamazepin, Rifampicin, Johanniskraut) |
3,98 |
20–60 |
|
Paliperidonpalmitat (PP3M) |
175–525/alle 12 W |
Unklar |
Unklar |
Umstellung von PP1M |
Alter, Nierenfunktionsstörung |
CYP3A4- und P-gp-Induktoren (z. B. Carbamazepin, Rifampicin, Johanniskraut) |
20–60 |
|
|
Risperidon Microspheres |
12,5–50/alle 2 W |
26 |
6 W |
21 Tage |
Alter, Leber- und Nierenfunktionsstörung, CYP2D6-PM |
CYP3A4- und P-gp-Induktoren (z. B. Carbamazepin), CYP2D6-Inhibitoren (Fluoxetin, Paroxetin) Alkohol, Levodopa, Cimetidin, Ranitidin, Clozapin |
5,392 |
20–60 |
|
Risperidon RBP-7000 |
90–120/alle 4 W |
9–11 |
3 Monate |
Nein |
CYP2D6-PM |
CYP2D6-Inhibitoren (Fluoxetin, Paroxetin) CYP3A4-Induktoren (Carbamazepin) |
20–60 |
|
|
Risperidon ISM |
75–100/alle 4 W |
7-11 |
Unklar |
Nein |
Alter, Leber- und Nierenfunktionsstörung |
CYP3A4- und P-gp-Induktoren (z. B. Carbamazepin), CYP2D6-Inhibitoren (Fluoxetin, Paroxetin) QTc-verlängernde Arzneimittel, Alkohol, Levodopa, Arzneimittel mit einem Hypotonierisiko, Psychostimulanzien |
2,68-3,57> |
20–60 |
1Für Aripiprazol gelten eine C/D und ein TRR für die aktive Wirkfraktion Aripiprazol + Dehydroaripiprazol von 21,89 bzw. 150–500 ng/ml.
2Der Wert gilt für die aktive Wirkfraktion. Der C/D Wert für die nicht metabolisierte Muttersubstanz Risperidon beträgt 0,57.
CYP: Cytochrom P450; C/D: dosisbezogene Konzentrationen oraler Darreichungsformen; PD: Pharmakodynamik; P-gp: P-Glykoprotein; PK: Pharmakokinetik; PM: Poor Metabolizer; UM: Ultrarapid Metabolizer; W: Woche(n)
Aripiprazol in verschiedenen Formulierungen
Aripiprazol-Monohydrat (AM), das nahezu unlöslich in Wasser ist, wird als lyophilisiertes Pulver formuliert. Nach Mischung mit Wasser bildet sich eine kristalline Suspension [81]. Die langsame Auflösung der Kristalle nach Injektion bewirkt eine verlängerte Absorptionszeit vom Muskel in den Blutkreislauf [44]. Die maximale Plasmakonzentration wird innerhalb von fünf bis sieben Tagen erreicht. Nach Injektion in den Musculus deltoideus lag die Plasmakonzentration um 31 % höher als nach einer Verabreichung in den Glutealmuskel. Die Unterschiede zwischen den Injektionsstellen sind nach Erreichen des Steady-States geringer. Nach der Injektion war der Verlauf der Plasmakonzentration von Dehydroaripiprazol (dem Hauptmetaboliten von Aripiprazol) fast parallel zum Verlauf der Aripiprazol-Plasmakonzentration [62].
Mit Aripiprazol-Lauroxil (AL) existiert zudem eine neue, in den USA zugelassene Aripiprazol-Prodrug-Depotformulierung [44]. Nach intramuskulärer Injektion wird AL langsam freigesetzt und durch Esterasen gespalten. Durch eine nichtenzymatische Hydrolyse entstehen aus einem Zwischenprodukt Aripiprazol und in geringen Mengen Formaldehyd, wobei Letztgenanntes leicht entgiftet wird [62]. Die Freisetzung von Aripiprazol in den Blutkreislauf erfolgt mit einer Verzögerung von fünf bis sechs Tagen und geschieht dann kontinuierlich über 36 Tage. Maximale Plasmakonzentrationen werden an Tag 41 nach deltoidaler Injektion erreicht [31, 94].
Olanzapinpamoat
In Olanzapinpamoat liegt Olanzapin als Salz der Pamoasäure vor, was die Löslichkeit des Moleküls verringert und dazu führt, dass der Wirkstoff nach der intramuskulären Injektion kontinuierlich freigesetzt wird [21, 37]. Das Salz ist nahezu wasserunlöslich, nach Mischung mit Wasser bildet sich eine Suspension. Nach einer Injektion in den Gesäßmuskel löst sich die Salzformulierung langsam auf, sodass Olanzapin und Pamoasäure in den Blutkreislauf gelangen [58]. Die langsame und gleichmäßige Auflösungsrate des Salzkomplexes führt zu einer kontinuierlichen Freisetzung von Olanzapin über eine Zeitraum von bis zu vier Wochen [76].
Paliperidon
Für Paliperidon existieren Depot-Formulierungen mit monatlichem, 3-monatigem und 6-monatigem Injektions-Intervall, wobei für letztere Applikationsform die Zulassung in den USA durch die FDA erst im September 2021 erfolgt ist. Die Einmonatsformulierung von Paliperidonpalmitat (PP1M) enthält Nanokristalle in einer wässrigen Suspension mit verzögerter Wirkstofffreisetzung [87]. Das 3-Monats-Paliperidonpalmitat (PP3M) unterscheidet sich von PP1M durch größere Nanokristallpartikel. Bei der PP3M-Formulierung sind diese noch größer, wodurch ein längeres Freisetzungsintervall begründet ist [87]. Nach der Injektion wird Paliperidonpalmitat durch Auflösung der Nanokristalle langsam freigesetzt. Anschließend erfolgt mittels Esterasen eine rasche Umwandlung zu Palmitinsäure und Paliperidon [76]. Die Freisetzung von Paliperidon in den systemischen Kreislauf wird hauptsächlich durch die langsame Auflösung der Nanokristalle gesteuert, welche wiederum von der Partikelgröße abhängig ist [15]. Die Paliperidon-Konzentrationen im Plasma erreichen bei PP1M nach 13 Tagen [15, 16] und bei PP3M nach 30 bis 33 Tagen ihr Maximum [37].
Risperidon in verschiedenen Formulierungen
Risperidon wird als sogenannte Mikrosphärenformulierung durch Verkapselung des Arzneimittels in biologisch abbaubare polymere Mikrokügelchen unter Verwendung von Poly-D,L-Lactid-co-Glycolid (PLGA) als Depot-Präparat hergestellt [24]. Es handelt sich um eine wässrige Suspension der Mikrosphären [32]. Die Mikrosphären werden nach Injektion allmählich hydrolysiert und Risperidon über einen Zeitraum von vier bis sechs Wochen freigesetzt [7, 32, 76].
Eine aktuelle Entwicklung, jedoch noch nicht in Deutschland verfügbar, ist Risperidon-RBP-7000. Diese Formulierung basiert auf einem biologisch abbaubaren In-situ-PLGA-Implantat, das im Gegensatz zu allen anderen verfügbaren Depot-Antipsychotika subkutan verabreicht wird [29]. Eine ebenfalls neue Depotformulierung ist Risperidon ISM (in situ-forming microparticle technology). Diese Technologie beinhaltet entweder eine Emulsion oder eine Suspension von Risperidon, die in einem festen polymeren Matrixsystem gebunden ist. Nach einer intramuskulären Injektion kommt es zu einer anhaltenden Wirkstofffreisetzung für bis zu einem Monat.
Pharmakokinetik der Depot-Antipsychotika im klinischen Alltag
Erkenntnisse zu den pharmakokinetischen Eigenschaften von Depot-Antipsychotika stammen im Wesentlichen aus drei Arten von Studien: 1) Studien in der Umstellungsphase von oraler Gabe auf ein Depot-Antipsychotikum; 2) Studien unter Depot-Behandlung, mit oder ohne Vergleichsgruppe mit oraler Medikation; 3) Studien mit kontinuierlichem TDM bei mit einem Depot behandelten Patienten, einschließlich erfolgter Untersuchungen beim Absetzen einer Depotmedikation.
Neben dem eigentlichen Arzneistoff zeigen auch die Metaboliten bei einer Reihe von Antipsychotika eine Aktivität und klinische Wirksamkeit. Die Erfassung der Wirkstoffkonzentrationen dieser aktiven Metaboliten ist daher von besonderer Relevanz, insbesondere, da sie Hinweise auf pharmakokinetische Wechselwirkungen und genetische Besonderheiten des Stoffwechsels liefern können. Unter den verfügbaren Depot-Antipsychotika sind hierzu Haloperidol, Risperidon und Aripiprazol am besten untersucht.
Ebenfalls aus pharmakokinetischer Sicht lohnend ist die Betrachtung des Verhältnisses zwischen Metaboliten und Muttersubstanz als sogenannter Metabolite-to-Parent-Ratio (MPR) vor und nach der Umstellung auf eine Depot-Formulierung.
TDM-gestützte Umstellung
Die Umstellung von einem oralen auf ein Depot-Antipsychotikum (oder umgekehrt) und der Wechsel zwischen verschiedenen Depot-Antipsychotika kommen im klinischen Alltag durchaus häufig vor. Immer mehr Studien widmen sich den damit verbundenen Fragestellungen, leider nur selten unter der Berücksichtigung von TDM-Daten oder pharmakokinetischen Aspekten [18, 66]. Daher ist die vorhandene Literatur, aus der Empfehlungen hinsichtlich eines begleitenden Wirkstoffspiegelmonitorings abgeleitet werden könnte, wenig aussagekräftig. Umfangreichere Untersuchungen liegen vornehmlich für Haloperidol und Risperidon in der Depotformulierung vor, weshalb diese beiden Substanze hier exemplarisch dargestellt werden.
Haloperidoldecanoat
Für Haloperidoldecanoat liegen umfangreiche pharmakokinetische Daten aus Studien zur Umstellungsphase vor [9, 19, 25, 45, 67, 97, 101]. Zielsetzung einiger dieser Untersuchungen war die Ermittlung eines TDM-basierten Umrechnungsfaktors zur Bestimmung der Depot-Äquivalenzdosis. Für diesen Faktor wurden, je nach Studie, Werte zwischen 9 und 30 ermittelt [9, 19, 25, 49]. Dabei ergibt sich die Dosis des Depots aus der Multiplikation der oralen Tagessdosis mit diesem Faktor. Die Umstellung erfolgte ausnahmslos ohne fortgeführte orale Haloperidol-Gabe.
In einer ersten Studie zur Umstellung auf Haloperidoldecanoat untersuchten Deberdt et al. Patientenkollektive, die auf eine monatliche Depot-Dosis entsprechend der 10-, 20- oder 30-fachen oralen Haloperidol-Tagesdosis umgestellt wurden. Die mittlere Wirkstoffkonzentration nach der ersten intramuskulären Gabe lag dabei im Vergleich höher (0,8–3,2 ng/ml) als nach oraler Gabe (< 2 ng/ml), bei einer Dosisbandbreite zwischen 30 und 300 mg alle vier Wochen für die Depotgabe. Nach der zweiten Gabe wurde ein Anstieg auf 2 bis 8 ng/ml gemessen, nach der dritten Injektion zeigte sich kein weiterer Anstieg, wobei dies nicht als Hinweis auf ein bereits eingetretenes Steady-State gewertet werden sollte.
In einer weiteren Studie wurden Patienten auf eine monatliche Haloperidoldecanoat-Dosis zwischen 75 bis 500 mg eingestellt. Daneben erhielten diese Patienten während der Umstellung überlappend für weitere zwei Wochen orale Haloperidol-Gaben [67, 97]. Für eine Patienten-Untergruppe (n = 22) wurden TDM-Daten erhoben. Die mittlere Wirkstoffkonzentration lag nach einer 2-wöchigen oralen Haloperidol-Behandlung (mittlere Haloperidol-Dosis 17 mg/Tag) bei 12,6 ng/ml und im Vergleich bei 7,49 ng/ml nach einer 2-monatigen Haloperidoldecanoat Behandlung, auch hier lag noch kein Steady-State vor. Unter Verwendung dieser Daten schlussfolgerten die Autoren einen Umrechnungsfaktor zwischen 9,4 und 15,0 zur Berechnung der Depot-Äquivalenzdosis.
Ereshefsky et al. [25] wählten eine alternative Vorgehensweise mit Verabreichung einer anfänglichen Loading-Dosis, die dem 20-Fachen der oralen Dosis entsprach. Im Verlauf wurde die Depot-Dosis jeden Monat um bis zu 25 % reduziert. In einer ersten Gruppe (n = 16) wurde von einer mittleren oralen Haloperidol-Dosis von 19 mg auf eine alle drei bis sieben Tage verabreichte Haloperidoldecanoat-Dosis zwischen 100 bis 200 mg umgestellt. In einer zweiten Gruppe (n = 8, mittlere orale Haloperidol-Dosis 40,6 mg/Tag) erfolgte die Umstellung auf 100 bis 150 mg Haloperidoldecanoat alle sieben bis 14 Tage, mit begleitender oraler Medikation. Eine dritte Gruppe (n = 3) erhielt eine monatliche Injektion von 100 mg Haloperidoldecanoat, bei vorausgegangener oraler Medikation mit durchschnittlich 33,6 mg/Tag Haloperidol, die nach erster Depot-Gabe nicht weiter fortgeführt wurde [25].
In der ersten Gruppe lag die mittlere Wirkstoffkonzentration nach 42 Tagen bei 8,5 ng/ml. Die zweite Gruppe wies in der Phase einer ausschließlich oralen Medikation Wirkstoffkonzentrationen von im Mittel 22,9 ng/ml auf. Unter der Depot-Gabe lag die gemessene Wirkstoffkonzentration in dieser Gruppe nie über 14,5 ng/ml. Nach Gabe einer einzigen 100-mg-Injektion (3. Gruppe) lag die Wirkstoffkonzentration im Mittel bei 19,7 ng/ml und war 28 Tage nach der Injektion auf 0,8 ng/ml abgefallen.
In einer weiteren Studie erhielten Patienten zunächst 100 mg Haloperidoldecanoat im wöchentlichen Intervall über insgesamt vier Wochen, dann alle zwei Wochen (ebenfalls vier Wochen lang), und schließlich einmal pro Monat, jeweils ohne ergänzende orale Gabe [45, 101]. Bei Patienten die zuvor 10 mg orales Haloperidol erhielten, lagen die Wirkstoffkonzentrationen vor Umstellung auf die Depot-Medikation bei etwa 8 ng/ml und sanken nach der Umstellung auf die Hälfte, ehe sie nach vier Injektionen innerhalb des ersten Monats allmählich wieder auf die anfänglich gemessenen Werte anstiegen [101]. Nach Verlängerung des Dosierungsintervalls auf monatliche Gaben blieb die Wirkstoffkonzentration auch über eine 52-wöchige Nachbeobachtungszeit unterhalb der initial unter oraler Gabe gemessenen Konzentrationen.
Aus einer Studie zu einer geriatrischen Population liegen Daten für elf Patienten vor, die initial 1 bis 9 mg orales Haloperidol erhielten und im Verlauf auf monatliche Haloperidoldecanoat-Gaben umgestellt wurden. Für die ersten zwei Injektionen wurde die 20-fache orale Dosis als Äquivalenz verabreicht, anschließend die 15-fache Dosis für die nächsten drei Injektionen. Unter oraler Medikation wurden initial Wirkstoffkonzentrationen von 1,5 ng/ml gemessen, diese stiegen nach der zweiten und dritten Injektion auf etwa 3,0 ng/ml [99].
Paliperidon
Zwei Studien lieferten TDM-Ergebnisse für Patienten, die von oralem Paliperidon auf monatliche Paliperidonpalmitat-Injektionen (PP1M) umgestellt wurden [64, 75]. Hierzu erhielten Patienten, die zuvor mit 6 mg oralem Paliperidon behandelt wurden, zunächst eine deltoidale PP1M-Injektion von 150 mg. Hiernach wurden sie randomisiert in Gruppen, die entweder 25, 100, 150 mg PP1M oder Placebo erhielten [75]. Nach der initialen PP1M-Gabe lagen die Wirkstoffkonzentrationen von Paliperidon bei 22,6 bis 23,3 ng/ml. In der mit 25 mg PP1M behandelten Gruppe sanken die Spiegel auf 11,5 ng/ml, stiegen allmählich auf 26,2 ng/ml an Tag 22 an und sanken an Tag 92 auf weniger als die Hälfte (10,2 ng/ml). Ein ähnliches Muster wurde in den 100- und 150-PP1M-Gruppen beobachtet, mit einem anfänglichen Abfall auf 13,3 ng/ml, gefolgt von einer Maximalkonzentration von 32,7 ng/ml an Tag 22 und einem Abfall auf 21,0 ng/ml an Tag 92 für die 100 mg sowie 13,9; 42,6; bzw. 28,4 ng/ml für die 150-mg-Gruppe.
Eine Phase-III-Studie mit einer dreimonatigen Formulierung von Paliperidonpalmitat (PP3M) lieferte TDM-Daten von Patienten, die von einer 17-wöchigen PP1M-Phase auf eine PP3M mit 3,5-facher Dosierung der PP1M-Injektion umgestellt wurden [14]. Aufgrund der gewählten Darstellungssystematik, der Fokussierung auf die klinischen Parameter Effektivität, Sicherheit und Zeit bis zum Rückfall und der mangelnden Kommentierung durch die Autoren lässt die Untersuchung aber keine Ableitung hinsichtlich erzielter Wirkstoffkonzentrationen unter der Dreimonatsdosierung von Paliperidonpalmitat zu.
TDM von Depot-Antipsychotika in der klinischen Routine
Indikationsstellung
Obwohl neuere Formulierungen von Depot-Antipsychotika der zweiten Generation herstellerseitig keine Überlappung mit einem oralen Antipsychotikum erfordern, scheint es klinisch geboten, eine orale Antipsychotika-Gabe im Sinne einer Cross-Titration überlappend mit der Depot-Eindosierung auszuschleichen [87]. Wesentliche Triebfeder dieser Empfehlung ist die sichere Umstellung eines oralen Antipsychotikums auf das wirkstoffgleiche Depot-Antipsychotikum mit Ausnahme der Umstellung von oralem Risperidon auf Paliperidonpalmitat oder von oralem Paliperidon auf Risperidon-Depot.
Eine TDM-geleitete Umstellung von einem oralen Antipsychotikum auf das wirkstoffgleiche Depotpräparat kann sich an folgendem Vorgehen orientieren (Abb. 2):
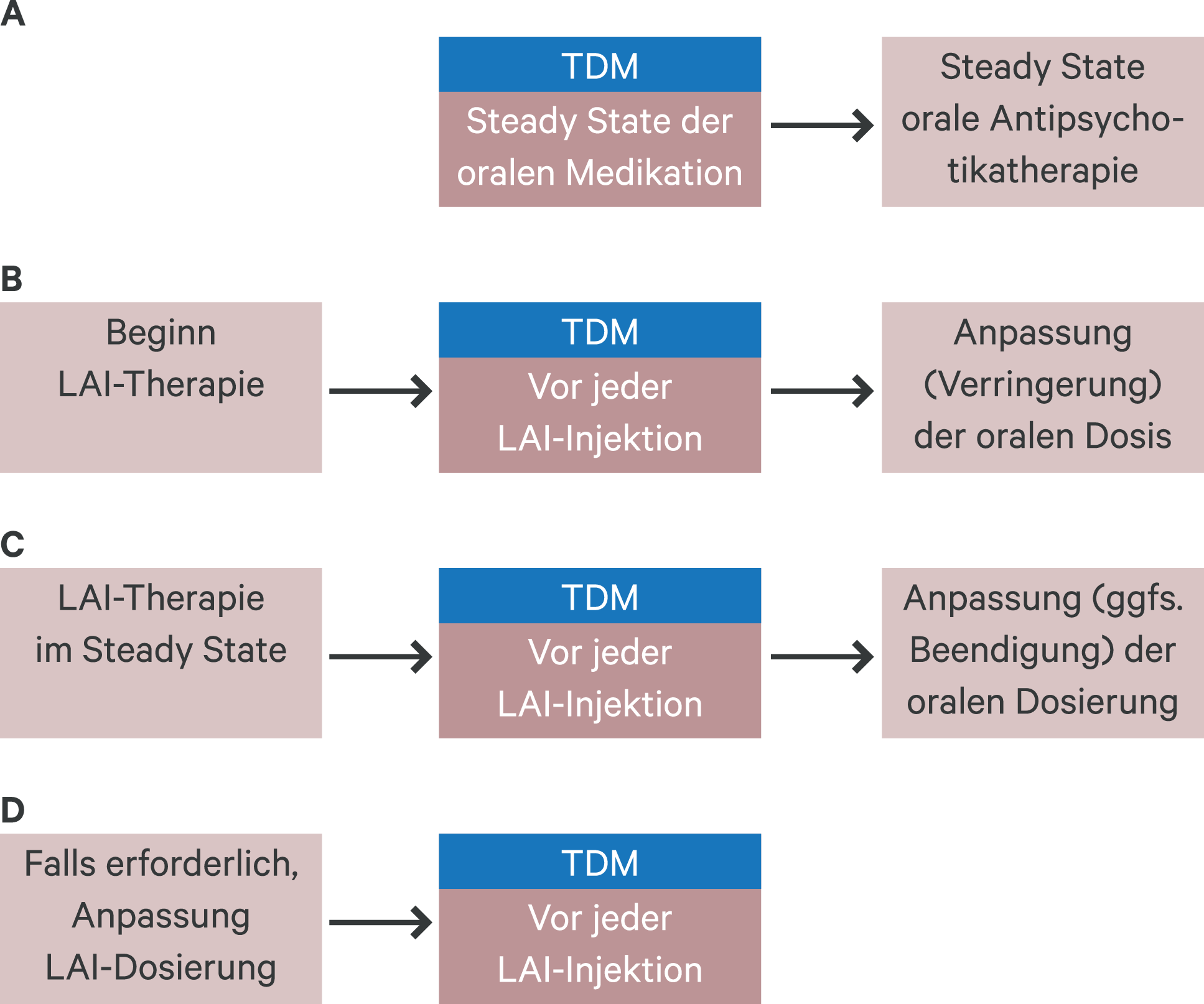
Abb. 2. Schematische Darstellung der Umstellung von oralen auf langwirksame Depot-Antipsychotika (LAI) und der Einsatz von therapeutischem Drug-Monitoring (TDM) in dieser Phase (nach [84])
Initiales TDM im Steady-State der oralen Medikation
Unter Steady-State-Bedingungen (nach 4–5 Halbwertszeiten) einer oralen Antipsychotika-Behandlung und bei Sicherstellung des Ansprechens gemäß klinischer Beurteilung auf die verordnete Substanz besteht ein erster vorbereitender Schritt auf eine Umstellung hin darin, TDM in Kombination mit der klinischen Einschätzung des Ansprechens auf die Behandlung einerseits und einer Bewertung der Verträglichkeit andererseits durchzuführen. Die Wirkstoffkonzentrationsmessung des oralen Antipsychotikums bei Ansprechen auf die Behandlung dient der Adhärenzkontrolle und gilt als Richtschnur für eine intendierte Depot-Behandlung. Informationen zur Adhärenz der Pharmakotherapie sind von wesentlicher Bedeutung, da Patienten mit Adhärenzproblemen eine wichtige Patientenuntergruppe für den Einsatz von Depot-Antipsychotika darstellen. Ein zusätzlicher klinischer Vorteil besteht in der Reduzierung von Risiken von unerwünschten Arzneimittelwirkungen, sollte es bei Umstellung auf ein Depot-Antipsychotikum zu einer abrupt hohen Antipsychotika-Exposition bei Patienten kommen, die ihre oralen Medikamente nur unzuverlässig eingenommen haben. Sollten mehrere Wirkstoffspiegelkontrollen erfolgt sein, könnte auch eine sorgsame Bewertung der intraindividuellen pharmakokinetischen Variabilität erfolgen. Die Bestimmung von Wirkstoffspiegelkonzentrationen unter der oralen antipsychotischen Medikation erlaubt eine präzise Dosisanpassung unter Berücksichtigung individueller pharmakokinetisch relevanter Eigenschaften wie Patientenalter, Geschlecht, genetische Besonderheiten, Raucherstatus oder Begleitmedikationen mit einer bekannten oder (bislang) unbekannten Einflussnahme auf den Metabolismus des Antipsychotikums.
TDM vor jeder Depot-Gabe zur Anpassung der oralen Begleitmedikation
Nach Beginn einer Behandlung mit einem Depot-Antipsychotikum kann die regelmäßige Anwendung von TDM unmittelbar vor der neuerlichen Depot-Injektion dazu beitragen, eine möglicherweise noch fortbestehende orale Dosierung schrittweise anzupassen bzw. gegebenenfalls reduzieren. Eine Ausnahme könnte eine Unverträglichkeit oder eine Ablehnung dosisveränderter Depot-Injektionen sein, sodass keine Veränderung der Depot-Dosierung erfolgt. In diesem Fall kann die Wirkstoffkonzentrationsmessung Einblicke in mögliche pharmakokinetische Korrelate aufgetretener Nebenwirkungen liefern. Dies ist besonders wichtig, da die Steuerungsfähigkeit bei Nebenwirkungen aufgrund der langen Wirkdauer der Depot-Antipsychotika häufig gering ist [43]. Angesichts bekannter intraindividueller Variabilitäten von Antipsychotika-Konzentrationen bleibt es jedoch mitunter herausfordernd, Veränderungen der Wirkstoffkonzentrationen richtig zu interpretieren. Darüber hinaus können Fluktuationen des Wirkstoffspiegels insbesondere für Antipsychotika mit größeren therapeutischen Breiten (große Spannweiten des therapeutischen Referenzbereichs) von geringerer klinischer Bedeutung sein [36].
TDM zur Dosisanpassung des Depot-Antipsychotikums
Nach dem Erreichen eines Steady-States für das entsprechende Antipsychotikum (vgl. Tab. 1, Tab. 2) kann TDM dazu genutzt werden, eine Dosisanpassung (oder das Absetzen) des oralen Antipsychotikums unter Berücksichtigung von Wirksamkeits- und Sicherheitsaspekten vorzunehmen [88]. Für den Fall, dass eine weitere Titration der Depot-Dosis erforderlich ist, kann das Vorgehen aus Schritt 2 wiederholt werden. Eine schematische Übersicht des klinikrelevanten Vorgehens zeigt Abbildung 2.
TDM in speziellen Situationen
Weitere Indikationen für TDM beim Gebrauch von Depot-Antipsychotika sind
- Rückfall oder Wiederauftreten von Symptomen
- Unerwünschte Arzneimittelwirkungen
- Kombination mit Arzneimitteln mit bekanntem pharmakokinetischem Interaktionspotenzial
- Genetischer Polymorphismus von CYP2D6 bei Depot-Antipsychotika, die über dieses Isoenzym verstoffwechselt werden (z. B. Aripiprazol oder Risperidon)
- Forensisch-psychiatrische Patienten
- Patienten mit akuten Entzündungen oder Infektionen
Praktische Empfehlung für die Durchführung von TDM bei Depot-Antipsychotika
Depot-Antipsychotika der ersten Generation (FGA)
Flupentixoldecanoat
Die Datenlage zu Flupentixol ist mehr als spärlich. Bildgebungsdaten/Rezeptorbesetzungsdaten (PET-Untersuchungen) liegen nur von zwei Patienten vor, die Flupentixol allerdings in Dosierungen erhielten, die vom Hersteller nicht mehr empfohlen werden. Die Erkenntnisse können daher nur als Anhaltspunkte angesehen werden. Bei den untersuchten Patienten mit monatlichen Flupentixol-Injektionen wurden Konzentrationen von mehr als 8 ng/ml erreicht. Diese waren mit einer sehr hohen Dopamin-D2-Rezeptor-Blockade verbunden, die oberhalb derjenigen lag, die innerhalb des derzeit gültigen therapeutischen Referenzbereich von 0,5–5 ng/ml zu erwarten ist. Unter Berücksichtigung von Daten aus Studien, in denen Dosierungen verwendet wurden, die den in der aktuellen klinischen Praxis angewandten nahe kamen [52], ist es wahrscheinlich so, dass sich der für orales Flupentixol vorgeschlagene Referenzbereich und der Bereich für die Depot-Applikation (0,5–5 ng/ml) sehr weit überlappen [36]. Der für orales Flupentixol geltende therapeutische Referenzbereich scheint insofern sehr gut übereinstimmend mit demjenigen Bereich zu sein, der anhand eines maximalen therapeutischen Nutzens für zweiwöchentliche Flupentixol-Dosen zwischen 20 und 40 mg im Steady-State zu erwarten ist [11]. Eine Verlängerung des Injektionsintervalls für den vorgesehenen Dosisbereich könnte entsprechend zu Spiegeln unterhalb des therapeutischen Referenzbereichs führen [40].
Fluphenazindecanoat
Verfügbare PET-Daten sind eher schwierig zu interpretieren, da bei allen Fluphenazindecanoat-behandelten Patienten die Dopamin-D2-Rezeptorbesetzung oberhalb des als therapeutisch sinnvoll angesehenen Bereichs lag und die verabreichten intramuskulären Dosierungen heute so nicht mehr verordnet werden. Die Autoren einer früheren Studie schlugen vor, die orale Dosis mit 2,5 zu multiplizieren, um die zweiwöchentliche Injektionsdosis abzuschätzen [51], während einer der Hersteller eine Äquivalenzdosierung einer 12,5-mg-Injektion alle drei Wochen mit einer oralen Tagesdosierung von 10 mg Fluphenazin vorschlug [8]. Ein beobachteter, rascher Abfall der Fluphenazin-Konzentrationen nach kurzer oder ausgesparter Umstellung unterstreicht die Notwendigkeit einer oralen Supplementation angesichts des sehr langsamen Anstiegs der Fluphenazin-Konzentrationen nach Injektion von Fluphenazindecanoat [69]. Andererseits kann der außergewöhnlich hohe Peak für Fluphenazin innerhalb von 24 Stunden nach der Injektion mit einem erhöhten Risiko für UAW gebunden sein, sodass der Kliniker innerhalb dieses Zeitfensters zusätzlich wachsam sein muss [51]. Verfügbare Steady-State-Daten von Patienten mit Fluphenazindecanoat zeigten oft Wirkstoffkonzentrationen unterhalb von 2 ng/ml, teilweise sogar unterhalb von 1 ng/ml, sodass der für orales Fluphenazin konstatierte Referenzbereich von 1 bis 10 ng/ml nach Umstellung auf ein Depot in den allermeisten Fällen wahrscheinlich nicht erreicht wird. Tatsächlich legen die verfügbaren Daten nahe, dass der empfohlene Referenzbereich der oralen Applikation zu hoch ist und die Obergrenze deutlich gesenkt werden sollte.
Fluspirilen
Für das aus der Gruppe der Diphenylbutylpiperidine stammende Depot-Antipsychotikum Fluspirilen existieren bislang keine validierten Daten über therapeutische Plasmaspiegel. Es wird ein Referenzbereich von 0,1 bis 2,2 ng/ml empfohlen [36], der sich auf zu erwartenden Konzentrationen im Serum oder Plasma bei Anwendung der empfohlenen Dosen bezieht. Daraufhin können evidenzbasierte Empfehlungen für den Einsatz von TDM nicht ausgesprochen werden.
Haloperidoldecanoat
Sehr niedrige Haloperidol-Konzentrationen in einem Bereich von 0,5 bis 3,5 ng/ml und damit unterhalb des therapeutischen Referenzbereichs für orale Dosierungen (1–10 ng/ml) [36] reichen wahrscheinlich aus, um eine Dopamin-D2-Rezeptorbesetzung innerhalb des gewünschten therapeutischen Fensters sicherzustellen [27, 68, 69]. Seit Durchführung eines europäischen Harmonisierungsprozesses sind die vormals sehr hohen oralen Dosierungen von Haloperidol obsolet. Vor dem Hintergrund vieler durchgeführter Studien mit den mittlerweile nicht mehr empfohlenen Depot-Dosierungen schlägt die US-amerikanische Arzneimittelbehörde FDA vor, dass anfänglich ausgewählte Haloperidol-Depot-Dosen 100 mg nicht überschreiten sollten [70]. Vorgeschlagen wird daher eine zunächst fortgesetzte orale Supplementierung unter Anwendung einer intramuskulären Depotformulierung, die das 10-Fache der oralen Dosis betragen sollte. Gleichzeitig empfiehlt sich hier dringend der Einsatz von TDM, welches für Haloperidol gemäß seinen Empfehlungskategorien als ohnehin „dringend empfohlen“ bewertet wird. Die orale Dosisanpassung sollte anhand der TDM-Befunde erfolgen, die im Um- und Einstellprozess auf die langwirksame Form erhalten wurden.
Bei älteren Patienten, bei denen es eher zu Akkumulationsphänomenen kommen könnte, kann eine schnellere Verringerung der oralen Dosen und eine zweiwöchentliche Anwendung von TDM empfohlen werden [99]. Dies gilt auch für Patienten mit bekanntem CYP2D6-„Poor-Metabolizer“(PM)-Status, bei denen eine niedrige Haloperidol-Verstoffwechselung berichtet wurde [74]. Einzelfallberichte weisen auf ein starkes Hemmpotenzial von Clozapin hinsichtlich des Metabolismus von Haloperidoldecanoat hin [6]. Aus diesem Grund scheint es empfehlenswert, bei der Kombination von Clozapin und Haloperidoldecanoat Vorsicht walten zu lassen.
Perphenazindecanoat und -enanthat
Zwischen den beiden langwirksamen Depotformulierungen von Perphenazin bestehen unter anderem pharmakokinetische Unterschiede dergestalt, dass bei Perphenazinenanthat höhere Wirkstofffluktuationen beobachtet wurden, die zu der höheren Prävalenz unerwünschter Arzneimittelwirkungen führen könnten [53].
Eine kleine PET-Studie zeigte für Perphenazindecanoat, dass bereits Konzentrationen von 1,1 bis 3 ng/ml zu einer Dopamin-D2-Rezeptorbesetzung von 65 bis 80 % führten. Hier zeigt sich ein deutlicher Überlappungsbereich mit dem empfohlenen Referenzbereich für orales Perphenazin (0,6–2,4 ng/ml) [36]. Da keine Transitionsuntersuchungen, also Erhebungen von Wirkstoffkonzentrationen im Übergang von oralem zu intramuskulärem Perphenazin vorliegen, bleibt eine Empfehlung hinsichtlich einer Initialdosierung der Depotform schwierig. Erkenntnisse aus Querschnittsdaten legen nahe, dass die orale Form von Perphenazin eine bis zu dreimal höhere Verstoffwechselung aufweist als die Depotformulierungen [92]. Konkrete Umstellungsstudien fehlen aber bisher, sodass nur die Empfehlung ausgesprochen werden kann, die Umstellung von oralem Perphenazin auf eine Depotformulierung unter begleitendem TDM durchzuführen. Ein weiterer Grund für die Empfehlung von TDM bei Perphenazin ist der Umstand, dass die Elimination von Perphenazindecanoat unabhängig von der Wirkstoffkonzentration erfolgt (sog. dosisunabhängige Kinetik), das heißt, der Konzentrationsabfall ist pro Zeit konstant [53]. Hierdurch besteht das potenzielle Risiko einer Wirkstoffakkumulation. Darüber hinaus gibt es Hinweise, die auf eine etwas höhere Verstoffwechselung bei Männern im Vergleich zu Frauen hindeuten. Die klinische Relevanz dieser geschlechtsspezifischen Unterschiede ist jedoch noch unklar.
Informationen über die pharmakokinetischen Eigenschaften von Perphenazinenanthat sind sehr begrenzt, was praktische Empfehlungen zum TDM stark erschwert.
Zuclopenthixoldecanoat und -acetat
Der empfohlene therapeutische Referenzbereich für orales Zuclopenthixol beträgt 4 bis 50 ng/ml [36]. Entsprechende Wirkstoffkonzentrationsmessungen bei verabreichtem Zuclopenthixoldecanoat zeigten im Mittel niedrigere Werte zwischen 4 und 20 ng/ml. Eine regelmäßige Anwendung von TDM scheint sinnvoll bei dem Versuch, eine wirksame Dosisauswahl vorzunehmen, verglichen mit einem starren Dosisalgorithmus nach Herstellervorgaben, wonach Patienten, die orale Tagesdosierungen von bis zu 20, 25 bis 40, 50 bis 75 und mehr als 75 mg erhalten, mit korrespondierenden zweiwöchentlichen Injektionen von 100, 200, 300 bzw. 400 mg behandelt werden sollten [61].
Da Zuclopenthixolacetat nicht für eine Erhaltungstherapie infrage kommt, empfehlen wir deshalb TDM nur zur Umstellung auf Zuclopenthixoldecanoat. Dennoch kann die Beurteilung von Zuclopenthixol-Wirkstoffkonzentrationen bei oral mit Zuclopenthixol behandelten Patienten als Alternative zum dosisbasierten Algorithmus des Herstellers angewandt werden, denn hiernach werden für orale Dosierungen von 20, 40 bzw. 60 mg starre Injektionen von 50, 100 und 150 mg Zuclopenthixolacetat vorgeschlagen [92].
Depot-Antipsychotika der zweiten Generation (SGA)
Aripiprazol in verschiedenen Formulierungen
Anders als für orales Aripiprazol existieren keine PET-Rezeptorbesetzungsdaten für entsprechende Depotformulierungen. Als partieller Agonist hängt die klinische Wirkung von Aripiprazol zudem weniger von der Bindungscharakteristik an den Rezeptoren ab, als dies bei anderen Antipsychotika der Fall ist. Als Folge können auch keine konsolidierten Empfehlungen für einen Referenzbereich eines langwirksamen Aripiprazol-Depots auf dieser Basis ausgesprochen werden.
Seitens des Herstellers wird eine Überlappung mit oralen Aripiprazol-Dosen von 10 bis 20 mg/Tag für zwei Wochen vorgeschlagen [72]. Diese Dosen führen erwartungsgemäß zu Aripiprazol-Plasmakonzentrationen in einem Bereich zwischen 81,0 und 300,0 ng/ml [36]. Eine naturalistische Untersuchung ergab, dass etwa ein Drittel gemessener Spiegel bei mit Aripiprazolmonohydrat behandelten Patienten unterhalb der unteren Grenze des therapeutischen Referenzbereichs von 100 ng/ml lagen [12]. Prospektive Daten zeigten jedoch, dass Werte unter 100 ng/ml nur bei Patienten beobachtet wurden, die Injektionen von 200 mg erhielten, was einer nicht zugelassenen Dosishöhe von Aripiprazol entspricht [62].
Die Auswertung verfügbarer Daten zu Wirkstoffbereichen von intramuskulär verabreichtem Aripiprazol scheinen den für orales Aripiprazol etablierten therapeutischen Referenzbereich zu stützen. Ein Bereich zwischen 100 bis 350 ng/ml scheint damit auch bei einer Behandlung mit einem Aripiprazol-Depot angemessen zu sein. Entsprechend gilt der vorgeschlagene Referenzbereich für die Summe aus Aripiprazol plus Dehydroaripiprazol von 150 bis 500 ng/ml [36, 85]. Da keine Daten aus Umstellungsstudien vorliegen, ist es nicht möglich, den Konzentrations-Zeit-Verlauf von Muttersubstanz und Metabolit während der Umstellphase vorherzusagen. Die Anwendung von TDM vor Beginn der Depotformulierung kann jedoch pharmakokinetische Phänotypen, beispielsweise von CYP2D6, aufdecken (aufgrund genetischer Besonderheiten oder einer Komedikation) und eine Behandlung mit veränderten Aripiprazol-Dosierungen und einer geplanten oralen Aripiprazol-Dosisanpassung speziell beispielsweise bei weiblichen CYP2D6-Langsammetabolisierern begleiten. Die Anwendung von TDM wird zudem bei Patienten über 65 Jahren empfohlen, die eher niedrigere Dosierungen benötigen, um eine Akkumulation zu vermeiden [95].
Der für Aripiprazol vorgeschlagene Referenzbereich könnte möglicherweise auch bei Patienten angewandt werden, die auf Aripiprazol-Lauroxil (AL) umgestellt wurden. Ergebnisse einer Single-Dose-Studie zu Aripiprazol-Lauroxil, also einer Einzelinjektionsuntersuchung des Ester-Prodrugs von Aripiprazol, zeigten einen Wirkstoffkonzentrationsbereich zwischen 113 und 133 ng/ml [65].
Der Hersteller empfiehlt eine orale Supplementierung bei der ersten Injektion in den Musculus deltoideus (nur 441 mg) oder in den Gesäßmuskel [80], gefolgt von Injektionen über vier (441 mg, 662 mg und 882 mg), sechs (882 mg) bzw. acht Wochen (1064 mg) [44]. Nur die Dosis von 441 mg AL ist für die deltoidale Injektion zugelassen, wobei die mittlere maximale Plasmakonzentration des Arzneimittels um 23 % höher ist als bei glutealer Injektion [94]. Nach einer ersten Injektion beträgt die Zeit bis zum Erreichen maximaler Plasmakonzentrationen 24,4 bis 35,2 Tage.
Olanzapinpamoat
In kleinen Kohortenstudien deuten PET-Daten darauf hin, dass die Injektion von 300 mg Olanzapinpamoat in einem vierwöchigen Intervall mit Wirkstoffkonzentrationen und einer korrespondierenden Dopamin-D2-Rezeptorblockade verbunden ist, die für viele Patienten zu niedrig erscheint. Diese Ergebnisse veranlassten den Hersteller, einen Dosierungsalgorithmus basierend auf oralen Olanzapin-Dosierungen vorzuschlagen. Hierbei werden Äquivalenzdosierungen zwischen 10 mg oralem Olanzapin und 210 mg alle zwei bzw. 405 mg alle vier Wochen intramuskulär angenommen. Patienten mit höheren oralen Tagesdosierungen (15–20 mg) sollten auf 300 mg intramuskulär alle zwei Wochen eingestellt werden [57]. Orale Tagesdosierungen zwischen 10 und 20 mg lassen Plasmakonzentrationen in einem Bereich von 11,9 bis 50,0 ng/ml erwarten. Patienten, die monatlich 405-mg-Injektionen erhielten, zeigten Wirkstoffkonzentrationen zwischen 8 bis 30 ng/ml im Steady-State, wobei höhere Konzentrationen nach Absetzen einer oralen Supplementierung wahrscheinlich nicht erreicht werden können [13, 63]. Bei Verwendung kürzerer Injektionsintervalle (z. B. 14-tägig) wurden höhere Konzentrationen von bis zu 40 ng/ml beobachtet [96]. Verfügbare Daten lassen daher annehmen, dass für die langwirksame intramuskuläre Injektionsform von Olanzapin andere therapeutische Referenzbereiche als für die orale Applikationsform gelten. Obwohl von großen Überschneidungen ausgegangen werden kann, scheinen effektive Wirkstoffkonzentrationen in einem Bereich zwischen 10 bis 40 ng/ml für Olanzapinpamoat zu liegen und damit deutlich niedriger als für orales Olanzapin, für das sich ein therapeutischer Referenzbereich zwischen 20 und 80 ng/ml etabliert hat.
Paliperidonpalmitat als monatliche (PP1M) oder dreimonatliche (PP3M) Depotformulierung
Auch für Paliperidonpalmitat liegen keine PET-Daten vor, die Auskünfte über dosis- bzw. konzentrationsabhänge Dopamin-D2-Rezeptorbesetzungen geben könnten. Ebenso existieren kaum TDM-Daten aus Umstellungsstudien. Insbesondere ist die Datenlage zu PP3M äußerst begrenzt, sodass keine allgemeinen Empfehlungen ausgesprochen werden können. Für die PP1M-Form existiert zwar eine größere Menge an TDM-Daten [87], hiervon entspricht jedoch nur ein sehr kleiner Teil wirklichen Steady-State-Bedingungen [12, 64, 104]. Gemessene mittlere Konzentrationen lagen in diesen Studien zwischen 28,9 und 40,3 ng/ml [87]. Der Hersteller schlägt eine tagesdosierungsabhängige Einstellung aus Paliperidon vor in der Art, dass Patienten, die 3 mg/Tag Paliperidon oral einnehmen, auf 25 bis 50 mg i. m., solche, die 6 mg/Tag einnehmen auf 75 mg i. m. sowie bei 9 mg/Tag auf 100 mg i. m. und bei 12 mg/Tag auf 150 mg i. m. umgestellt werden sollten [86]. Allerdings scheint dieser Algorithmus aufgrund von Resorptionsunterschieden zwischen oralem Paliperidon und der Darreichung als Depotformulierung von begrenztem Nutzen zu sein [87]. Patienten, die mit niedrigeren oralen Paliperidon-Dosen (< 4 mg/Tag) behandelt werden, zeigten ein gewisses Risiko für eine abrupt auftretende, hohe Paliperidon-Exposition nach Umstellung auf PP1M, da durchaus Paliperidon-Konzentrationen oberhalb von 20 ng/ml zu erwarten sind. Bei Patienten mit oralen Paliperidon-Dosen über 5 mg/Tag kann die regelmäßige Anwendung von TDM eine Auswahl der Depotdosierung ermöglichen. Angesichts berichteter Schwierigkeiten bei einer überlappungsfreien Umstellung empfehlen wir einen an TDM-Befunden orientierten Umstieg von oralem Paliperidon hin zu PP1M. Auch die Umstellung von oralem Risperidon sollte entsprechend dem vorgestellten Vorgehen geschehen [39]. Orale Tagesdosierungen von Risperidon von mehr als 4 mg für mindestens eine Woche scheinen gemäß dem beschriebenen Vorgehen problemlos umstellbar zu sein [39]. Eine Dosisäquivalenzumstellung von Patienten, die 4, 5 und 6 mg Risperidon oral erhielten, wurde mit 100, 100 bis 150 bzw. 150 mg PP1M vorgeschlagen [87]. Fallberichte weisen darauf hin, dass der Steady-State in einigen Patienten erst nach längerer Behandlungszeit mit PP1M erreicht wird (Abb. 3).
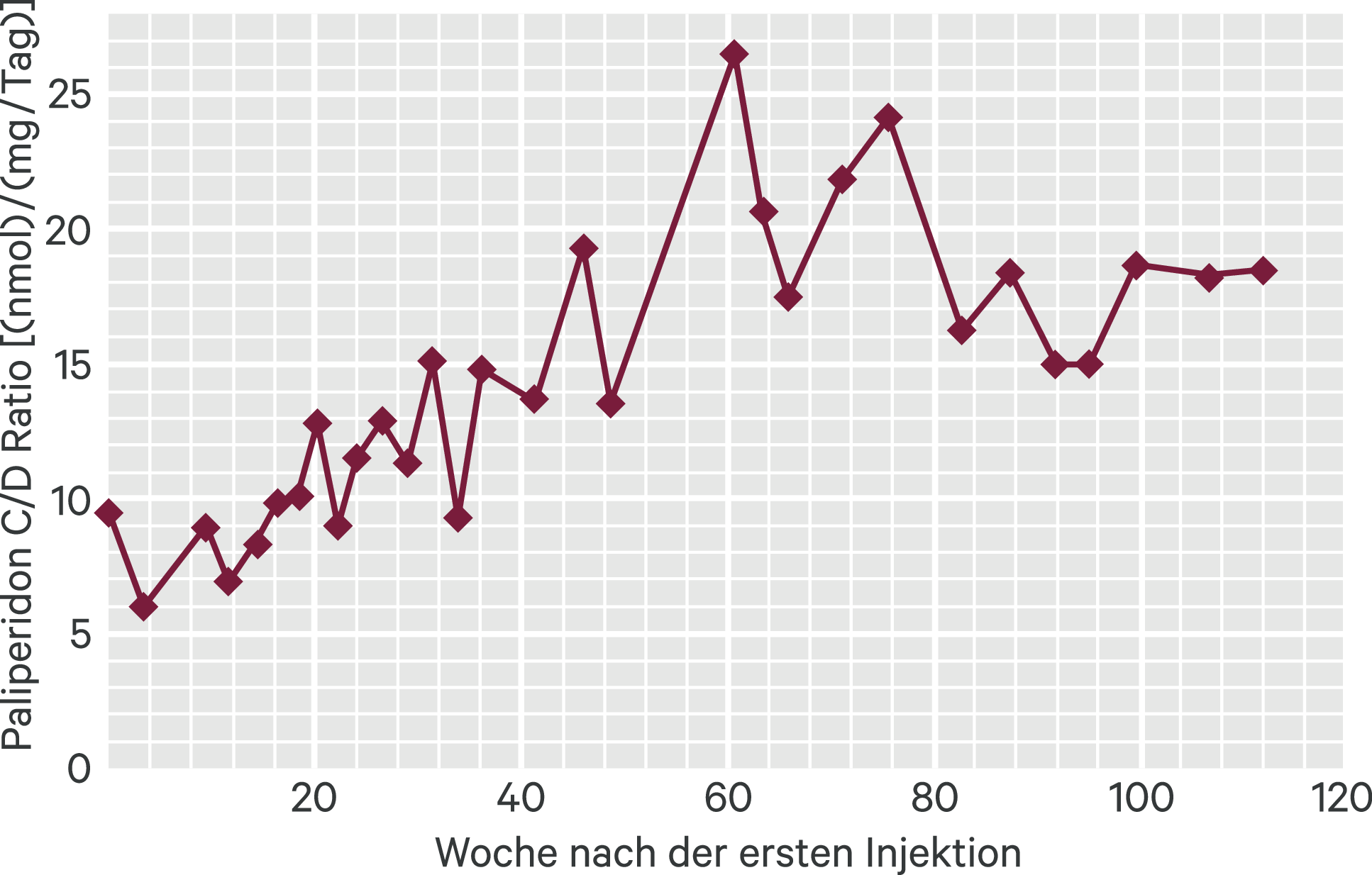
Abb. 3. 30 nacheinander bestimmte dosiskorrigierte Wirkstoffkonzentrationen (C/D) von Paliperidon bei einem 29-jährigen Mann, der mit einer intramuskulären Gabe von Paliperidon PP1M über einen Zeitraum von mehr als zwei Jahren behandelt wurde. Die Dosierung lag ab Woche 26 konstant bei 150 mg alle 17 Tage. Zwölf nach Woche 26 bestimmte Wirkstoffkonzentrationen ließen erst ab etwa Woche 56 Steady-State-Bedingungen annehmen. Hier lag der mittlere C/D-Quotient bei 19,3 ± 3,6 [(nmol/l)/(mg/d)]. Anmerkung: Umrechnung von nmol/l in ng/ml mittels CF (Conversion Factor) = 2,35. Es gilt: (CF) nmol/l = ng/ml × CF (eigene Darstellung nach [35])
Geriatrische Patienten oder Patienten mit einer bestehenden Niereninsuffizienz benötigen aufgrund der renalen Ausscheidung von Paliperidon offensichtlich niedrigere Dosierungen [35]. Andererseits zeigen Patienten mit einer Komedikation mit Carbamazepin trotz der nur sehr geringgradig ausgeprägten hepatischen Metabolisierung von Paliperidon niedrigere Paliperidon-Spiegel und Dosisanpassungen erscheinen erforderlich, sobald Carbamazepin gleichzeitig angewandt wird [35]. Zudem wiesen Patienten, die genotypisch als CYP2D6-Ultrarapid-Metabolizer (UM) identifiziert wurden, etwa 50 % niedrigere dosiskorrigierte Wirkstoffkonzentrationen (C/D) auf als die Normalmetabolisierer, während die Poor Metabolizer etwa 30 % höhere C/D-Werte aufwiesen [59]. Dementsprechend sollten – falls vorhanden – Kenntnisse des Metabolisiererstatus in die Entscheidung über die Dosishöhe von PP1M mit einfließen.
Für PP3M schlägt der Hersteller einen dosisorientierten Algorithmus vor, wonach Patienten, die 25, 50, 100 und 150 mg PP1M für mindestens 120 Tage erhalten haben, auf 175, 350, 450 bzw. 525 mg PP3M umzustellen sind [87]. Mangels vorhandener TDM-Daten kann aus dieser Warte keine Empfehlung ausgesprochen werden. Noch schwieriger scheint es, vorherzusagen, wie sich die Äquivalenzdosierungen von PP3M im Steady-State verhalten.
Risperidon Microspheres und RBP-7000
Die sogenannten Risperidon-Microsphere-Formulierungen zeigen in PET-Studien mit oralem und Risperidon-Depot eine gute Übereinstimmung zwischen den Formulierungen. Aus den verschiedenen Untersuchungen lässt sich ein Schwellenwert von etwa 10 bis 15 ng/ml für ein optimales therapeutisches Ansprechen ableiten. Wirkstoffkonzentrationen zwischen 40 und 50 ng/ml zeigen tendenziell höhere Inzidenzen extrapyramidal-motorischer Nebenwirkungen. In einer vorherigen Übersichtsarbeit konnten wir zeigen, dass die mittleren Konzentrationen der aktiven Wirkfraktion (Risperidon + Hydroxyrisperidon) unter Steady-State-Bedingungen zwischen 22,5 und 44,3 ng/ml lagen [86]. Basierend auf diesen Daten scheint der therapeutische Referenzbereich der aktiven Wirkfraktion oral verabreichten Risperidons (20–60 ng/ml) eine sehr gute Orientierung zu geben, obwohl nur eine geringe Anzahl von Patienten diesen Bereich im Steady-State zu erreichen scheint [40].
In der Zulassungsstudie erhielten Patienten, die mit 2, 4 oder 6 mg/Tag oralem Risperidon stabilisiert waren, im zweiwöchentlichen Abstand eine 25- oder 50-mg-Injektion der Risperidon-Microspheres mit oraler Ergänzung für zwei Wochen. Hier konnte nach erfolgter erster Injektion die Dosierung der zweiten Injektion halbiert werden [23]. Neben einer Dosistitration empfehlen wir auch TDM für Patienten, die mit Risperidon-Microspheres behandelt werden, wenn sie beispielsweise zwischen einer Deltoidal- und Glutealinjektion wechseln, da die Resorption zunehmen kann, sobald Patienten eine deltoidale Injektion erhalten. Wenn zudem CYP2D6-Genotypisierungsdaten verfügbar sind, können niedrigere Dosierungen angewandt werden, die durch intensives TDM begleitet werden, da bei Langsammetabolisierern eine deutlich reduzierte Clearance vorliegt. Ebenso besteht eine reduzierte Risperidon-Clearance bei Patienten mit Niereninsuffizienz, sodass auch hier insbesondere eine TDM-geleitete Pharmakotherapie erfolgen sollte.
Für Risperidon RBP-7000 empfiehlt die Fachinformation, dass Patienten, die mit 3 mg und 4 mg oralem Risperidon behandelt wurden, auf monatliche Injektionen mit 90 mg bzw. 120 mg umgestellt werden können. Gleichzeitig empfiehlt die Fachinformation, dass Patienten, die mehr als 4 mg oder weniger als 3 mg Risperidon oral erhalten, keine Kandidaten für eine Umstellung auf RBP-7000 seien [41]. Da die klinische Erfahrung jedoch noch sehr begrenzt ist und die Daten aus pharmakokinetischen Modellanalysen stammen, können derzeit keine Empfehlungen ausgesprochen werden [29].
Fazit
Die moderne Psychopharmakotherapie verfolgt das Ziel einer individualisierten und personalisierten Therapieoptimierung, ohne dass hierzu ein breites Instrumentarium zur Verfügung steht, das es erlaubt, Arzneimitteltherapiesicherheit und Therapieeffektivität patientenzentriert zu gestalten. Aufgrund der langen Verweildauer von Depotformulierungen im Körper gilt es hier insbesondere, pharmakokinetische Besonderheiten zu berücksichtigen und die Therapie mit TDM als evidenzbasierte Methode sowohl zur Effektivitätsverbesserung als auch zur Förderung der Therapiesicherheit zu begleiten. Hierdurch ist es in besonderem Maße möglich, die interindividuellen pharmakokinetischen Variabilitäten zu adressieren und damit eine möglichst nebenwirkungsfreie Behandlung sicherzustellen. Daher sollte die erfolgreiche Integration von TDM in die klinische Praxis auch bei der Anwendung von Depot-Formulierungen fester Bestandteil in einer erfolgreichen Therapiegestaltung sein. Dies gilt insbesondere bei der Umstellung von oralen Antipsychotika auf Depotformulierungen, da die pharmakokinetischen Erkenntnisse über orale Formulierungen von Antipsychotika nicht 1 : 1 auf Depotformulierungen zu übertragen sind [43]. Die vorliegende Übersicht zeigt, dass für einzelne Depotformulierungen möglicherweise andere, oftmals niedrigere therapeutische Referenzbereiche existieren. Gleichwohl scheint das Wissen hierüber meist noch von anekdotischer Evidenz zu sein und bedarf der weiteren sorgsamen klinischen Bewertung.
Die Anwendung von TDM anstelle von Dosisäquivalenzalgorithmen bei der Umstellung erlaubt eine bessere Annäherung an die im Rahmen der Langzeitbehandlung oftmals angestrebte minimal effektive Dosis bzw. die minimal effektive Wirkstoffkonzentration. Aufgrund der oftmals langen Dauer bis zum Erreichen eines stabilen pharmakokinetischen Gleichgewichts, dem Steady-State, scheint TDM insbesondere hier therapieoptimierend einsetzbar zu sein, um das Erreichen eines stabilen Gleichgewichts laborchemisch begleiten zu können und den Kliniker auch darauf hinzuweisen, wann dieser Zustand dann ohne große intraindividuellen Schwankungen der Wirkstoffkonzentrationen erreicht ist.
Sobald ein Steady-State erreicht ist, hilft TDM dabei, die Therapie stabil und trotz äußerer Einflüsse kontrolliert zu gestalten, wenn beispielsweise pharmakokinetische Einflussnahmen im Rahmen von (neu begonnener) Polymedikation zu möglichen Veränderungen der Konzentrationen der Depot-Antipsychotika führen könnten. Patienten mit im Laufe der psychischen Grunderkrankung oftmals hinzutretenden somatischen Begleiterkrankungen, die auch zu einer veränderten Arzneistoffelimination führen können (z. B. eine eingeschränkte Nierenfunktion), können einer Akkumulation des Antipsychotikums und damit einhergehenden toxischen Phänomenen stärker ausgesetzt sein. Hier sollte dann TDM auch von Depot-Antipsychotika ein fester Bestandteil der Psychopharmakotherapie sein. Dies gilt auch für ältere Patienten, bei denen altersbedingte physiologische Veränderungen zu einer verminderten Nierenfunktion bzw. veränderten Verstoffwechselung von Medikamenten führen können. Bei Patienten, die mit mehreren Arzneimitteln derselben oder unterschiedlicher pharmakologischer Klassen behandelt werden, das heißt bei bestehender Multimedikation inklusive Depot-Antipsychotika, ist TDM zur Aufklärung von Arzneimittelwechselwirkungen von wesentlicher Bedeutung. Damit bleibt festzuhalten, dass mit TDM – auch wenn das Wissen bei Depot-Antipsychotika teilweise noch spärlich ist – ein effektives klinisches Instrument zur Verbesserung von Sicherheit und Wirksamkeit einer Behandlung mit einem langwirksamen Depot-Antipsychotikum zur Verfügung steht. Insbesondere gehört hierzu ein sorgfältiges Monitoren der Verträglichkeit der verschriebenen Depotmedikation mit regelmäßigen TDM-Kontrollen. Dieses aktive klinische Management sollte gemeinsam mit dem aktiven Einbezug der Patienten in den Entscheidungsprozess erfolgen, um mögliche Vorbehalte der Patienten gegenüber einem Depot-Antipsychotikum zu reduzieren und die Adhärenz zu verbessern.
Interessenkonflikterklärung
Priv.-Doz. Dr. Paulzen erhielt 2020/2021 Honorare für Vorträge und Beratungsleistungen von Lundbeck, Neuraxpharm, Janssen Cilag, Novartis und Otsuka. Er ist Herausgeber von PSIAC, einem internetbasierten Interaktionsprogramm mit der Schwerpunktsetzung auf Psychopharmaka. Ein Interessenkonflikt bezüglich des Beitrags besteht nicht. Dr. Schoretsanitis erhielt 20/2021 Honorare für Beratungsleistungen von HLS Therapeutics. Dr. Liebe berichtet keinen Interessenkonflikt. Prof. Hiemke erhielt 2020/2021 Honorare für Vorträge von Otsuka.
Literatur
1. Aaes-Jørgensen T. Pharmacokinetics of three different injectable zuclopenthixol preparations. Prog Psychopharmacol Biol Psychiatry 1989;13:77–85.
2. Abdel-Baki A, Thibault D, Medrano S, et al. Long-acting antipsychotic medication as first-line treatment of first-episode psychosis with comorbid substance use disorder. Early Interv Psychiatry 2020;14:69–79.
3. Abhijnhan A, Adams CE, David A, et al. Depot fluspirilene for schizophrenia. Cochrane Database Syst Rev 2007;(1):CD001718.
4. Abrams R, Swartz CM. Electroconvulsive therapy and prolactin release: relation to treatment response in melancholia. Convuls Ther 1985;1:38–42.
5. Alastanos JN, Paxos C, Emshoff J. Evaluation of oral antipsychotic supplementation of select second-generation long-acting injectable antipsychotics in an acute-care psychiatric setting. Ment Health Clin 2019;9:18–23.
6. Allen SA. Effect of chlorpromazine and clozapine on plasma concentrations of haloperidol in a patient with schizophrenia. J Clin Pharmacol 2000;40:1296–7.
7. Altamura AC, Sassella F, Santini A, et al. Intramuscular preparations of antipsychotics: uses and relevance in clinical practice. Drugs 2003;63:493–512.
8. APP Pharmaceuticals LLC. Fluphenazine decanoate. Schaumburg, IL2010.
9. Aymard N, Viala A, Stein I, et al. Pharmacoclinical correlations in schizophrenic patients treated with haloperidol decanoate: clinical evaluations, concentrations of plasma and red blood cell haloperidol and its reduced metabolite, and plasma homovanillic acid. Prog Neuropsychopharmacol Biol Psychiatry 1995;19:1119–35.
10. Baastrup PC, Alhfors UG, Bjerkenstedt L, et al. A controlled Nordic multicentre study of zuclopenthixol acetate in oil solution, haloperidol and zuclopenthixol in the treatment of acute psychosis. Acta Psychiatr Scand 1993;87:48–58.
11. Bailey L, Taylor D. Estimating the optimal dose of flupentixol decanoate in the maintenance treatment of schizophrenia – a systematic review of the literature. Psychopharmacology (Berl) 2019;236:3081–92.
12. Baldelli S, Clementi E, Cattaneo D. Can we rely on AGNP therapeutic targets also for lai antipsychotics? Pharmacopsychiatry 2018;51:270–1.
13. Baldelli S, Mauri MC, Di Pace C, et al. Intraindividual and interindividual variability of olanzapine trough concentrations in patients treated with the long-acting injectable formulation. J Clin Psychopharmacol 2018;38:365–9.
14. Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial. JAMA Psychiatry 2015;72:830–9.
15. Chue P, Chue J. A review of paliperidone palmitate. Expert Rev Neurother 2012;12:1383–97.
16. Citrome L. New second-generation long-acting injectable antipsychotics for the treatment of schizophrenia. Expert Rev Neurother 2013;13:767–83.
17. Coutinho E, Fenton M, Quraishi S. Zuclopenthixol decanoate for schizophrenia and other serious mental illnesses. Cochrane Database Syst Rev 2000;(2):CD001164.
18. Covell NH, McEvoy JP, Schooler NR, et al. Effectiveness of switching from long-acting injectable fluphenazine or haloperidol decanoate to long-acting injectable risperidone microspheres: an open-label, randomized controlled trial. J Clin Psychiatry 2012;73:669–75.
19. Deberdt R, Elens P, Berghmans W, et al. Intramuscular haloperidol decanoate for neuroleptic maintenance therapy. Efficacy, dosage schedule and plasma levels. An open multicenter study. Acta Psychiatr Scand 1980;62:356–63.
20. Detke HC, Zhao F, Garhyan P, et al. Dose correspondence between olanzapine long-acting injection and oral olanzapine: recommendations for switching. Int Clin Psychopharmacol 2011;26:35–42.
21. Di Lorenzo R, Brogli A. Profile of olanzapine long-acting injection for the maintenance treatment of adult patients with schizophrenia. Neuropsychiatr Dis Treat 2010;6:573–81.
22. Diaz-Fernandez S, Frias-Ortiz DF, Fernandez-Miranda JJ. Suicide attempts in people with schizophrenia before and after participating in an intensive case managed community program: a 20-year follow-up. Psychiatry Res 2020;287:112479.
23. Eerdekens M, Van Hove I, Remmerie B, et al. Pharmacokinetics and tolerability of long-acting risperidone in schizophrenia. Schizophr Res 2004;70:91–100.
24. Ereshefsky L, Mannaert E. Pharmacokinetic profile and clinical efficacy of long-acting risperidone: potential benefits of combining an atypical antipsychotic and a new delivery system. Drugs in R&D 2005;6:129–37.
25. Ereshefsky L, Toney G, Saklad SR, et al. A loading-dose strategy for converting from oral to depot haloperidol. Hosp Community Psychiatry 1993;44:1155–61.
26. Fagiolini A, Alfonsi E, Amodeo G, et al. Switching long acting antipsychotic medications to aripiprazole long acting once-a-month: expert consensus by a panel of Italian and Spanish psychiatrists. Expert Opin Drug Saf 2016;15:449–55.
27. Farde L, Suhara T, Nyberg S, et al. A PET-study of [11C]FLB 457 binding to extrastriatal D2-dopamine receptors in healthy subjects and antipsychotic drug-treated patients. Psychopharmacology (Berl) 1997;133:396–404.
28. Fluanxol®Depot (Flupenthixol Decanoate Intramuscular Injection), 2017.
29. Gomeni R, Heidbreder C, Fudala PJ, et al. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol 2013;53:1010–9.
30. Gopal S, Gassmann-Mayer C, Palumbo J, et al. Practical guidance for dosing and switching paliperidone palmitate treatment in patients with schizophrenia. Curr Med Res Opin 2010;26:377–87.
31. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol 2018;38:435–41.
32. Harrison TS, Goa KL. Long-acting risperidone: a review of its use in schizophrenia. CNS Drugs 2004;18:113–32.
33. Hata T, Kanazawa T, Hamada T, et al. The 12-year trend report of antipsychotic usage in a nationwide claims database derived from four million people in Japan. J Psychiatr Res 2020;127:28–34.
34. Hefner G, Laux G, Baumann P, et al. Konsensus-Leitlinien für therapeutisches Drug-Monitoring in der Neuropsychopharmakologie: Update 2017. Psychopharmakotherapie 2018;25:92–140.
35. Helland A, Spigset O. Serum concentrations of paliperidone after administration of the long-acting injectable formulation. Ther Drug Monit 2017;39:659–62.
36. Hiemke C, Bergemann N, Clement HW, et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: update 2017. Pharmacopsychiatry 2018;51:9–62.
37. Hope JD, Keks NA. Paliperidone palmitate three-month depot formulation: a helpful innovation with practical pitfalls. Australas Psychiatry 2018;26:206–9.
38. Howes OD, McCutcheon R, Agid O, et al. Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on diagnosis and terminology. Am J Psychiatry 2017;174:216–29.
39. Hsia SL, Leckband SG, Rao S, et al. Dosing strategies for switching from oral risperidone to paliperidone palmitate: effects on clinical outcomes. Ment Health Clin 2017;7:95–100.
40. Hýža M, Šilhán P, Češková E, et al. Plasma levels of long-acting injectable antipsychotics in outpatient care: a retrospective analysis. Neuropsychiatr Dis Treat 2021;17:1069–75.
41. Indivior. Perseris® (risperidone) for extended-release injectable suspension, for subcutaneous use. Slough, UK2018.
42. Jaen JC, Caprathe BW, Pugsley TA, et al. Evaluation of the effects of the enantiomers of reduced haloperidol, azaperol, and related 4-amino-1-arylbutanols on dopamine and sigma receptors. J Med Chem 1993;36:3929–36.
43. Jang S, Woo J. Five month-persistent extrapyramidal symptoms following a single injection of paliperidone palmitate: a case report. Clin Psychopharmacol Neurosci 2017;15:288–91.
44. Jann MW, Penzak SR. Long-acting injectable second-generation antipsychotics: an update and comparison between agents. CNS Drugs 2018;32:241–57.
45. Jann MW, Wei FC, Lin HN, et al. Haloperidol and reduced haloperidol plasma concentrations after a loading dose regimen with haloperidol decanoate. Prog Neuropsychopharmacol Biol Psychiatry 1996;20:73–86.
46. Janzen D, Bolton J, Kuo IF, et al. Trends in the use of long-acting injectable antipsychotics in the province of Manitoba, Canada. J Clin Psychopharmacol 2020;40:6–13.
47. Jayakody K, Gibson RC, Kumar A, et al. Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses. Cochrane Database Syst Rev 2012;(4):CD000525.
48. Jorgensen A. Pharmacokinetic studies on flupenthixol decanoate, a depot neuroleptic of the thioxanthene group. Drug Metab Rev 1978;8:235–49.
49. Kane JM. Dosage strategies with long-acting injectable neuroleptics, including haloperidol decanoate. J Clin Psychopharmacol 1986;6:20S–3S.
50. Kim HO, Seo GH, Lee BC. Real-world effectiveness of long-acting injections for reducing recurrent hospitalizations in patients with schizophrenia. Ann Gen Psychiatry 2020;19:1.
51. Kissling W, Möller HJ, Walter K, et al. Double-blind comparison of haloperidol decanoate and fluphenazine decanoate effectiveness, side-effects, dosage and serum levels during a six months’ treatment for relapse prevention. Pharmacopsychiatry 1985;18:240–5.
52. Kistrup K, Gerlach J, Aaes-Jorgensen T, et al. Perphenazine decanoate and cis(z)-flupentixol decanoate in maintenance treatment of schizophrenic outpatients. Serum levels at the minimum effective dose. Psychopharmacology (Berl) 1991;105:42–8.
53. Knudsen P, Hansen LB, Larsen NE. Perphenazine decanoate in sesame oil vs. perphenazine enanthate in sesame oil: a comparative study of pharmacokinetic properties and some clinical implications. Acta Psychiatr Scand Suppl 1985;322:11–4.
54. Lahteenvuo M, Tanskanen A, Taipale H, et al. Real-world effectiveness of pharmacologic treatments for the prevention of rehospitalization in a Finnish nationwide cohort of patients with bipolar disorder. JAMA Psychiatry 2018;75:347–55.
55. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia – a critical systematic review and meta-analysis of randomised long-term trials. Schizophr Res 2011;127:83–92.
56. Li P, Snyder GL, Vanover KE. Dopamine targeting drugs for the treatment of schizophrenia: past, present and future. Curr Top Med Chem 2016;16:3385–403.
57. Lilly USA. Zyprexa Relprevv® (olanzapine) for extended release injectable suspension. Indianapolis, IN2012.
58. Lindenmayer JP. Long-acting injectable antipsychotics: focus on olanzapine pamoate. Neuropsychiatr Dis Treat 2010;6:261–7.
59. Lisbeth P, Vincent H, Kristof M, et al. Genotype and co-medication dependent CYP2D6 metabolic activity: effects on serum concentrations of aripiprazole, haloperidol, risperidone, paliperidone and zuclopenthixol. Eur J Clin Pharmacol 2016;72:175–84.
60. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serious mental illness. BMC Psychiatry 2013;13:340.
60a. Lohse MJ. Psychopharmaka. In: Schwabe U, Ludwig WD (Hrsg). Arzneiverordnungs-Report 2020. Berlin, Heidelberg: Springer, 2020: 781–814.
61. Lundbeck. Clopixol®depot (200 mg/ml Zuclopenthixol Intramuscular Injection). St-Laurent, QC2016.
62. Mallikaarjun S, Kane JM, Bricmont P, et al. Pharmacokinetics, tolerability and safety of aripiprazole once-monthly in adult schizophrenia: an open-label, parallel-arm, multiple-dose study. Schizophr Res 2013;150:281–8.
63. Mauri MC, Maffini M, Di Pace C, et al. “Long-acting” olanzapine in maintenance therapy of schizophrenia: a study with plasma levels. Int J Psychiatry Clin Pract 2015;19:99–105.
64. Mauri MC, Paletta S, Di Pace C, et al. Paliperidone long-acting plasma level monitoring and a new method of evaluation of clinical stability. Pharmacopsychiatry 2017;50:145–51.
65. Mauri MC, Reggiori A, Minutillo A, et al. Paliperidone LAI and aripiprazole LAI plasma level monitoring in the prophylaxis of bipolar disorder type i with manic predominance. Pharmacopsychiatry 2020;53:209–19.
66. Möller HJ, Llorca PM, Sacchetti E, et al. Efficacy and safety of direct transition to risperidone long-acting injectable in patients treated with various antipsychotic therapies. Int Clin Psychopharmacol 2005;20:121–30.
67. Nayak RK, Doose DR, Nair NP. The bioavailability and pharmacokinetics of oral and depot intramuscular haloperidol in schizophrenic patients. J Clin Pharmacol 1987;27:144–50.
68. Nyberg S, Farde L, Bartfai A, et al. Central D2 receptor occupancy and effects of zuclopenthixol acetate in humans. Int Clin Psychopharmacol 1995;10:221–7.
69. Nyberg S, Farde L, Halldin C. Delayed normalization of central D2 dopamine receptor availability after discontinuation of haloperidol decanoate. Preliminary findings. Arch Gen Psychiatry 1997;54:953–8.
70. Ortho-McNeil-Janssen Pharmaceuticals. Haldol® (haloperidol) Decanoate Intramuscular Injection. Titusville, NJ2011.
71. Ostuzzi G, Mazzi MA, Terlizzi S, et al. Factors associated with first- versus second-generation long-acting antipsychotics prescribed under ordinary clinical practice in Italy. PLoS One 2018;13:e0201371.
72. Otsuka. Abilify Maintena® (aripiprazole) for extended-release injectable suspension, for intramuscular use. Tokyo, Japan, 2016.
73. Pan L, Belpaire FM. In vitro study on the involvement of CYP1A2, CYP2D6 and CYP3A4 in the metabolism of haloperidol and reduced haloperidol. Eur J Clin Pharmacol 1999;55:599–604.
74. Panagiotidis G, Arthur HW, Lindh JD, et al. Depot haloperidol treatment in outpatients with schizophrenia on monotherapy: impact of CYP2D6 polymorphism on pharmacokinetics and treatment outcome. Ther Drug Monit 2007;29:417–22.
75. Pandina GJ, Lindenmayer JP, Lull J, et al. A randomized, placebo-controlled study to assess the efficacy and safety of 3 doses of paliperidone palmitate in adults with acutely exacerbated schizophrenia. J Clin Psychopharmacol 2010;30:235–44.
76. Park EJ, Amatya S, Kim MS, et al. Long-acting injectable formulations of antipsychotic drugs for the treatment of schizophrenia. Arch Pharm Res 2013;36:651–9.
77. Park SC, Oh HS, Tripathi A, et al. Cannabis use correlates with aggressive behavior and long-acting injectable antipsychotic treatment in Asian patients with schizophrenia. Nord J Psychiatry 2019;73:323–30.
78. Peitl V, Badzim VA, Sisko Markos I, Rendulic A, et al. Improvements of frontotemporal cerebral blood flow and cognitive functioning in patients with first episode of schizophrenia treated with long-acting aripiprazole. J Clin Psychopharmacol 2021;41:638–43.
79. Quraishi S, David A. Depot flupenthixol decanoate for schizophrenia or other similar psychotic disorders. Cochrane Database Syst Rev 2000;(2):CD001470.
80. Raedler LA. Aripiprazole lauroxil (Aristada): long-acting atypical antipsychotic injection approved for the treatment of patients with schizophrenia. Am Health Drug Benefits 2016;9:40–3.
81. Rauch AS, Fleischhacker WW. Long-acting injectable formulations of new-generation antipsychotics: a review from a clinical perspective. CNS Drugs 2013;27:637–52.
82. Remenar JF. Making the leap from daily oral dosing to long-acting injectables: lessons from the antipsychotics. Mol Pharm 2014;11:1739–49.
83. Samtani MN, Gopal S, Gassmann-Mayer C, et al. Dosing and switching strategies for paliperidone palmitate: based on population pharmacokinetic modelling and clinical trial data. CNS Drugs 2011;25:829–45.
84. Schoretsanitis G, Baumann P, Conca A, et al. Therapeutic drug monitoring of long-acting injectable antipsychotic drugs. Ther Drug Monit 2021;43:79–102.
85. Schoretsanitis G, Kane JM, Correll CU, et al. Blood levels to optimize antipsychotic treatment in clinical practice: a joint consensus statement of the American Society of Clinical Psychopharmacology and the Therapeutic Drug Monitoring Task Force of the Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie. J Clin Psychiatry 2020;81:19cs13169.
86. Schoretsanitis G, Spina E, Hiemke C, et al. A systematic review and combined analysis of therapeutic drug monitoring studies for long-acting risperidone. Expert Rev Clin Pharmacol 2017;10:965–81.
87. Schoretsanitis G, Spina E, Hiemke C, et al. A systematic review and combined analysis of therapeutic drug monitoring studies for long-acting paliperidone. Expert Rev Clin Pharmacol 2018;11:1237–53.
88. Schoretsanitis G, Villasante-Tezanos AG, de Leon J. Studies of half-lives of long-acting antipsychotics are needed. Pharmacopsychiatry 2019;52:45–6.
89. Sheehan JJ, Reilly KR, Fu D-J, et al. Comparison of the peak-to-trough fluctuation in plasma concentration of long-acting injectable antipsychotics and their oral equivalents. Innov Clin Neurosci 2012;9:17–23.
90. Stauning JA, Kirk L, Joorgensen A. Comparison of serum levels after intramuscular injections of 2 % and 10 % cis(Z)-flupentixol decanoate in Viscoleo to schizophrenic patients. Psychopharmacology (Berl) 1979;65:69–72.
91. Szukalski B, Lipska B, Welbel L, et al. Serum levels and clinical response in long-term pharmacotherapy with zuclopenthixol decanoate. Psychopharmacology (Berl) 1986;89:428–31.
92. Thomsen K, Olesen OV, Poulsen JH. [The effect of serum monitoring on concentration of perphenazine and zuclopenthixole]. Ugeskr Laeger 1993;155:2443–8.
93. Tuninger E, Levander S. Large variations of plasma levels during maintenance treatment with depot neuroleptics. Br J Psychiatry 1996;169:618–21.
94. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic, following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res 2014;159:404–10.
95. Tveito M, Molden E, Hoiseth G, et al. Impact of age and CYP2D6 genetics on exposure of aripiprazole and dehydroaripiprazole in patients using long-acting injectable versus oral formulation: relevance of poor and intermediate metabolizer status. Eur J Clin Pharmacol 2020;76:41–9.
96. Tveito M, Smith RL, Molden E, et al. Age impacts olanzapine exposure differently during use of oral versus long-acting injectable formulations: an observational study including 8,288 patients. J Clin Psychopharmacol 2018;38:570–6.
97. Vasavan Nair NP, Suranyi-Cadotte B, Schwartz G, et al. A clinical trial comparing intramuscular haloperidol decanoate and oral haloperidol in chronic schizophrenic patients: efficacy, safety, and dosage equivalence. J Clin Psychopharmacol 1986;6:30S–7S.
98. Viala A, Ba B, Durand A, et al. Comparative study of the pharmacokinetics of zuclopenthixol decanoate and fluphenazine decanoate. Psychopharmacology (Berl) 1988;94:293–7.
99. Viukari M, Salo H, Lamminsivu U, et al. Tolerance and serum levels of haloperidol during parenteral and oral haloperidol treatment in geriatric patients. Acta Psychiatr Scand 1982;65:301–8.
100. Voirol P, Jonzier-Perey M, Porchet F, et al. Cytochrome P-450 activities in human and rat brain microsomes. Brain Res 2000;855:235–43.
101. Wei FC, Jann MW, Lin HN, et al. A practical loading dose method for converting schizophrenic patients from oral to depot haloperidol therapy. J Clin Psychiatry 1996;57:298–302.
102. Wistedt B, Ranta J. Comparative double-blind study of flupenthixol decanoate and fluphenazine decanoate in the treatment of patients relapsing in a schizophrenic symptomatology. Acta Psychiatr Scand 1983;67:378–88.
103. Yazdi K, Rosenleitner J, Pischinger B. [Combination of two depot antipsychotic drugs]. Nervenarzt 2014;85:870–1.
104. Zipursky Z, Huynh H, Agid O, et al. Can long-acting injectable paliperidone dosing be optimized with plasma levels measurements? Schizophr Bull 2019;44:S415.
*Geteilte Autorschaft
Dr. med. Dr. phil. Georgios Schoretsanitis, Psychiatrische Universitätsklinik Zürich (PUK), Lenggstrasse 31, 8008 Zürich, Schweiz, und Department of Psychiatry, Zucker Hillside Hospital, Northwell Health, Glen Oaks, New York, USA, E-Mail: george.schor@gmail.com
Priv.-Doz. Dr. med. Michael Paulzen, Alexianer Krankenhaus Aachen, Alexianergraben 33, 52062 Aachen, und Klinik für Psychiatrie, Psychotherapie und Psychosomatik, Medizinische Fakultät, RWTH Aachen
Dr. med. Claus Liebe, Alexianer Krankenhaus Aachen, Alexianergraben 33, 52062 Aachen
Prof. Dr. Christoph Hiemke, Klinik für Psychiatrie und Psychotherapie, Universitätsmedizin der Johannes Gutenberg-Universität Mainz, Untere Zahlbacher Straße 8, 55131 Mainz
Clinical routine therapeutic drug monitoring for long-acting injectable antipsychotics
Originally developed for chemical restraint, long-acting injectable antipsychotics (LAIs) received over the years a completely different role in modern psychopharmacology with various benefits in the treatment of patients with schizophrenia spectrum disorders and other severe mental illnesses. Accordingly, prescription rates for LAIs grow constantly pointing out the strong need for tools to improve LAI-based treatment safety and efficacy outcomes in clinical practice. Therapeutic drug monitoring (TDM) is a clinical tool with long tradition consisting of the measurement and the interpretation of drug levels in the blood of patients; TDM evidence is quite robust in the treatment with oral antipsychotics, whereas for LAIs current knowledge is less advanced. A recent review work focusing on TDM as regular part of LAI-treatment assessed available literature to provide suggestions for the efficient TDM integration in clinical practice with LAIs. Ultimate goal of establishing TDM in the treatment with LAIs is to improve everyday aspects of efficacy, e.g. LAI dose selection optimization and safety, e.g. assessment/management of LAI-associated adverse reactions. A common principle in TDM for LAIs is the adoption of the therapeutic reference ranges that have been suggested for their oral counterparts. However, there are hints that in some cases lower antipsychotic levels in LAI-treated patients may be associated with therapeutic efficacy. Future research should address the need for more precise LAI therapeutic reference ranges, which might slightly differ from reference ranges for oral antipsychotics. Despite the need for TDM science advances, TDM remains a unique tool to account for individual patients’ characteristics and their impact on drug bioavailability in titration and long-term treatment with LAIs.
Key words: Long-acting injectable antipsychotics (LAIs); therapeutic drug monitoring (TDM); schizophrenia spectrum disorders; psychopharmacology; personalized medicine
Psychopharmakotherapie 2022; 29(01):2-16