Holger Petri, Bad Wildungen*
Proteinkinase-Inhibitoren als „Victim Drugs“
Die Phase-I-Reaktionen im Fremdstoffmetabolismus werden primär über Cytochrom-P450(CYP)-Enzyme katalysiert. Von diesen hat das Isoenzym CYP3A4 den größten Einfluss. In den Fachinformationen der Proteinkinase-Inhibitoren („Victim Drugs“) finden sich Angaben zum Abbau der einzelnen Arzneimittel in Abhängigkeit von CYP3A4-Modulatoren („Perpetrator Drugs“), die zumeist aus klinischen Studien an Probanden gewonnen wurden. Dabei werden Modulatoren mit maximaler Effektstärke auf das jeweilige Enzym eingesetzt. Hierdurch wird ein Worst-Case-Szenario simuliert. Als CYP3A4-Hemmer werden bevorzugt Azol-Antimykotika wie Itraconazol und Ketoconazol verwendet, als CYP3A4-Induktor Rifampicin.
In den Tabellen 2 und 3 sind die Veränderungen der AUC(Fläche unter der Konzentrations-Zeit-Kurve)-Werte aufgeführt, die mit solchen starken CYP3A4-Modulatoren in klinischen Studien gewonnen wurden [1, 4].
Tab. 2. AUC-Werterhöhungen der Proteinkinase-Inhibitoren durch starke CYP3A4-Inhibitoren
|
AUC-Werterhöhung |
Ibrutinib (24,0-fach), Everolimus (15,3-fach), Midostaurin (10,0-fach), |
|
AUC-Werterhöhung |
Dasatinib, Lapatinib, Ribociclib, Crizotinib, Nilotinib, Ceritinib, Temsirolimus, Axitinib |
|
AUC-Werterhöhung |
Ruxolitinib, Palbociclib, Erlotinib, Gefitinib, Ponatinib, Alectinib, Dabrafenib, Pazopanib, Sunitinib, Imatinib, Cabozantinib |
|
Keine signifikanten Änderungen |
Afatinib, Lenvatinib, Nintedanib, Osimertinib, Sorafenib, Tivozanib, Trametinib, Vandetanib, Vemurafenib |
Angaben ohne Berücksichtigung der Metaboliten (Ausnahme Temsirolimus i. v.), Aufzählung mit abnehmender AUC-Werterhöhung
Tab. 3. AUC-Senkungen der Proteinkinase-Inhibitoren durch starke CYP3A4-Induktoren
|
AUC-Senkung ≥ 80 % |
Midostaurin (96 %), Bosutinib (94 %), Ibrutinib (90 %), Ribociclib (89 %), Palbociclib (85 %), Crizotinib (84 %), Gefitinib (83 %), Dasatinib (82 %), Nilotinib (80 %) |
|
AUC-Senkung ≥ 50 % bis < 80 % |
Axitinib, Osimertinib, Cabozantinib, Alectinib, Lapatinib, Ceritinib, Imatinib, Ruxolitinib, Everolimus, Ponatinib, Erlotinib, Temsirolimus, Nintedanib |
|
AUC-Senkung ≥ 20 % bis < 50 % |
Tivozanib, Sunitinib, Vandetanib, Vemurafenib, Sorafenib, Dabrafenib, Afatinib |
|
Keine signifikanten Änderungen |
Lenvatinib, Trametinib |
Aufzählung mit abnehmender AUC-Senkung; Cobimetinib und Pazopanib wurden in vivo nicht untersucht, wahrscheinlich kommt es zu einer relevanten AUC-Senkung
Andere CYP-Enzyme sind nur vereinzelt von Bedeutung im Stoffwechsel der Proteinkinase-Inhibitoren (Tab. 1 im Online-Supplement zu diesem Beitrag). Dabrafenib wird neben CYP3A4 auch über CYP2C8 abgebaut. Der starke CYP2C8-Hemmer Gemfibrozil steigerte die AUC von Dabrafenib um 47 % [4]. Inhaltsstoffe des Tabakrauchs induzieren die Bildung von CYP1A2. Das Rauchen von Zigaretten erniedrigte die Exposition gegenüber Erlotinib um 50 bis 60 % [4]. Ciprofloxacin, ein mittelstarker CYP1A2-Inhibitor, erhöhte den AUC-Wert von Erlotinib um 39 % [4]. Vorsicht ist geboten, wenn Ciprofloxacin oder starke CYP1A2-Inhibitoren wie Fluvoxamin komediziert werden. Gefitinib ist auch Substrat von CYP2D6. Bei Patienten mit vermindertem CYP2D6-Stoffwechsel (z. B. Poor-Metabolizer) kann die Behandlung mit einem wirksamen CYP3A4-Inhibitor zu höheren Gefitinib-Konzentrationen führen. Dies sollte auch bei Patienten mit extensivem Stoffwechsel beachtet werden, wenn der Patient starke CYP2D6-Hemmer einnimmt wie Bupropion, Fluoxetin oder Paroxetin. Bei Ruxolitinib ist die Einzeldosis bei Kombination mit dualen Inhibitoren von CYP2C9 und CYP3A4 wie Fluconazol um etwa die Hälfte zu reduzieren [4].
Proteinkinase-Inhibitoren als „Perpetrator Drugs“
Die Proteinkinase-Inhibitoren können selbst die Exposition anderer Arzneimittel verändern. Als CYP3A4-Testsubstrat dient oral eingenommenes Midazolam. Der AUC-Wert des Benzodiazepins erhöhte sich durch Crizotinib um das 3,7-Fache, durch Imatinib um das 3,5-Fache, durch Nilotinib um das 2,6-Fache und durch Ribociclib dosisabhängig um das 3,8- bis 5,2-Fache.
Geringfügig stieg die systemische Verfügbarkeit des CYP2D6-Testsubstrates Metoprolol durch Gefitinib und Imatinib um 35 % bzw. 23 %. In einer ähnlichen Größenordnung führte die CYP2C8-bedingte Hemmung durch Lapatinib und Pazopanib zu einem AUC-Anstieg von Paclitaxel (23 % bzw. 25 %).
Dagegen ist die Steigerung der Plasmaspiegel des CYP1A2-Testsubstrats Tizanidin durch Vemurafenib um das 4,7-Fache von klinischer Relevanz. Vorsicht ist geboten in Kombination mit Arzneimitteln mit geringer therapeutischer Breite, deren Abbau von diesem Enzym maßgeblich abhängig ist, beispielsweise Agomelatin, Clozapin und Theopyhllin.
Fazit
- Von den aktuell in Deutschland verfügbaren Proteinkinase-Inhibitoren sind mit Ausnahme von Temsirolimus alle oral einzunehmen. Daraus ergeben sich gegenüber der parenteralen Gabe zusätzliche Variablen, die Einfluss auf das Ausmaß der Wirkstoffexposition haben und zur hohen inter- und intraindividuellen Variabilität mancher Proteinkinase-Inhibitoren beitragen können. Diese umfassen neben CYP-bedingten Interaktionen unter anderem die gleichzeitige Nahrungsaufnahme, der Einfluss des Magen-pH-Werts, die Affinität zu intestinalen und hepatischen Arzneimitteltransportern und die präsystemische Biotransformation [5].
- Die Daten aus den zulassungsrelevanten Interaktionsstudien sind nicht immer ohne Weiteres auf die meist multimorbiden älteren Patienten übertragbar. Beispielsweise erhöhten starke CYP3A4-Inhibitoren die Midostaurin-Exposition bei Patienten nur um das 1,4- bis 2,1-Fache, was zu einer vergleichbaren Verträglichkeit mit der Placebo-Kontrollgruppe führte [2, 4]. Imatinib hemmt seinen eigenen Metabolismus (Autoinhibition). Unter Steady-State-Bedingungen führte der starke CYP3A4-Hemmer Ritonavir zu keiner signifikanten Änderung der Imatinib-Exposition [6].
- Angaben in den Fachinformationen stammen häufig aus In-vitro-Untersuchungen und bestätigen sich nicht bei der Anwendung am Menschen. Beispielsweise ist Regorafenib (Stivarga ist in Deutschland vom Markt genommen und nur noch als Einzelimport zu beziehen) in vitro in Konzentrationen, die im Steady State erreicht werden können, ein Inhibitor von CYP2C8, 2C9, 2C19 und 3A4. In einer klinischen Studie zeigte sich jedoch, dass die Abbauhemmung der Testsubstrate durch Regorafenib ohne klinische Relevanz blieb [4].
- In Ermangelung klinisch-pharmakokinetisch dokumentierter Fallserien zu den meisten Proteinkinase-Inhibitoren sollten Verordner kontinuierlich die Fachpublikationen auf diesbezügliche Studienveröffentlichungen und Kasuistiken sichten. So lange sind nach Möglichkeit klinisch relevante Modulatoren, soweit in der Fachinformation gefordert, zu meiden.
- Bei der Abschätzung der Wechselwirkungsrisiken der meist komplexen Therapieschemata unterstützen Interaktionsdatenbanken. Risikoärmere therapeutische Alternativen können der Literatur zu diesem Thema entnommen werden [3].
- Schwer abschätzbar wird es, wenn bei der peroralen Applikation von Onkologika das Problem der Einnahme von teilweise hochdosierten Natur- und Nahrungssupplementen durch den Patienten hinzukommt.
- Bei nicht vermeidbaren Risikokombinationen sollten, soweit hierfür Referenzbereiche definiert sind, Dosisanpassungen auch mit Unterstützung des therapeutischen Drug-Monitorings (TDM) durchgeführt werden.
- Treten schwerwiegende Nebenwirkungen mutmaßlich durch Wechselwirkungen auf, sind Ärzte und Apotheker nach Berufsordnung angehalten, den Fall zu dokumentieren und beispielsweise an die Arzneimittelkommission der Deutschen Ärzteschaft (AkdÄ) zu melden. So wird ein wichtiger Beitrag zur Arzneimitteltherapiesicherheit (AMTS) geleistet.
- Die Praxistipps sollten auch bei Neueinführung von weiteren Proteinkinase-Inhibitoren Anwendung finden.
Praxistipps bei Kombination mit CYP3A4-Modulatoren
- Nach Absetzen eines CYP3A4-Hemmers ist eine Auswaschphase zu beachten, bevor der Proteinkinase-Inhibitor wieder in der gleichen Dosis gegeben wird wie vor der gleichzeitigen Behandlung. Diese beträgt mindestens drei bis fünf Halbwertszeiten, bei den meisten Wirkstoffen etwa zwei bis drei Tage (Ausnahme: z. B. Amiodaron und irreversibel wirkende Inhibitoren).
- Auch vom Konsum von Grapefruitsaft wird häufig abgeraten, Ibrutinib darf nicht mit Grapefruitsaft und Bitterorangensaft eingenommen werden. Die Zitrusfrüchte enthalten CYP3A4-hemmende Inhaltsstoffe [8].
- Schwerwiegende UAW drohen bei Wirkstoffen mit QTc-Zeit-verlängernden Eigenschaften wie Bosutinib und Nilotinib. Ein Risikofaktor für die Entwicklung von Torsade-de-pointes-Arrhythmien sind erhöhte Spitzenplasmaspiegel [7]. Zudem verlängern einige CYP3A4-Inhibitoren wie die Makrolid-Antibiotika Clarithromycin und Erythromycin selbst die QTc-Zeit [9].
- Die Effekte des Induktors treten mit zeitlichem Verzug von ein bis zwei Wochen auf und halten nach Absetzen etwa so lange an (Deinduktionszeit).
- Johanniskraut als starker CYP3A4-Induktor ist mit einigen Proteinkinase-Inhibitoren kontraindiziert (z. B. Ibrutinib, Midostaurin, Tivozanib).
- Abbildung 1 enthält eine Auswahl klinisch relevanter CYP3A4-Modulatoren.
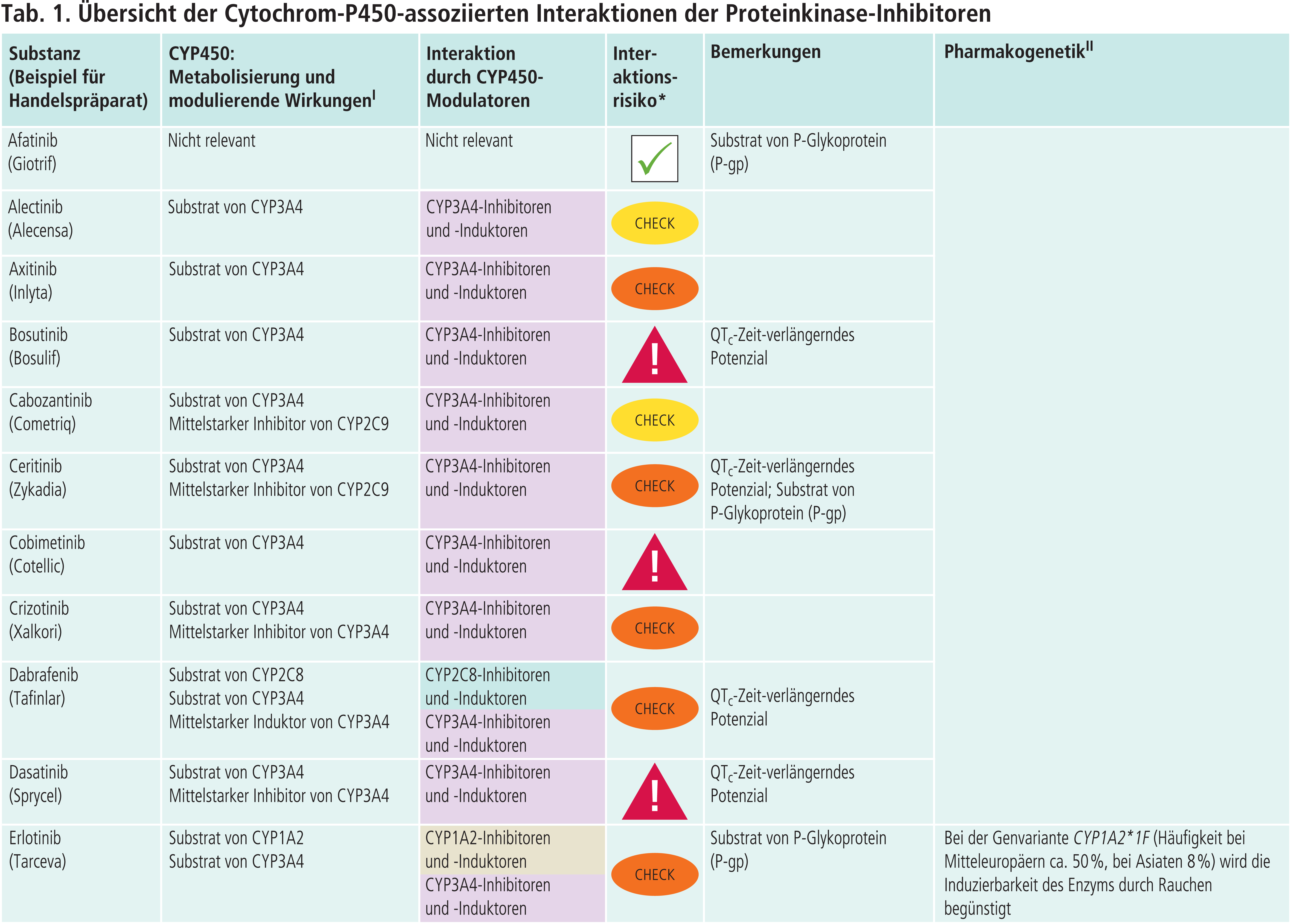
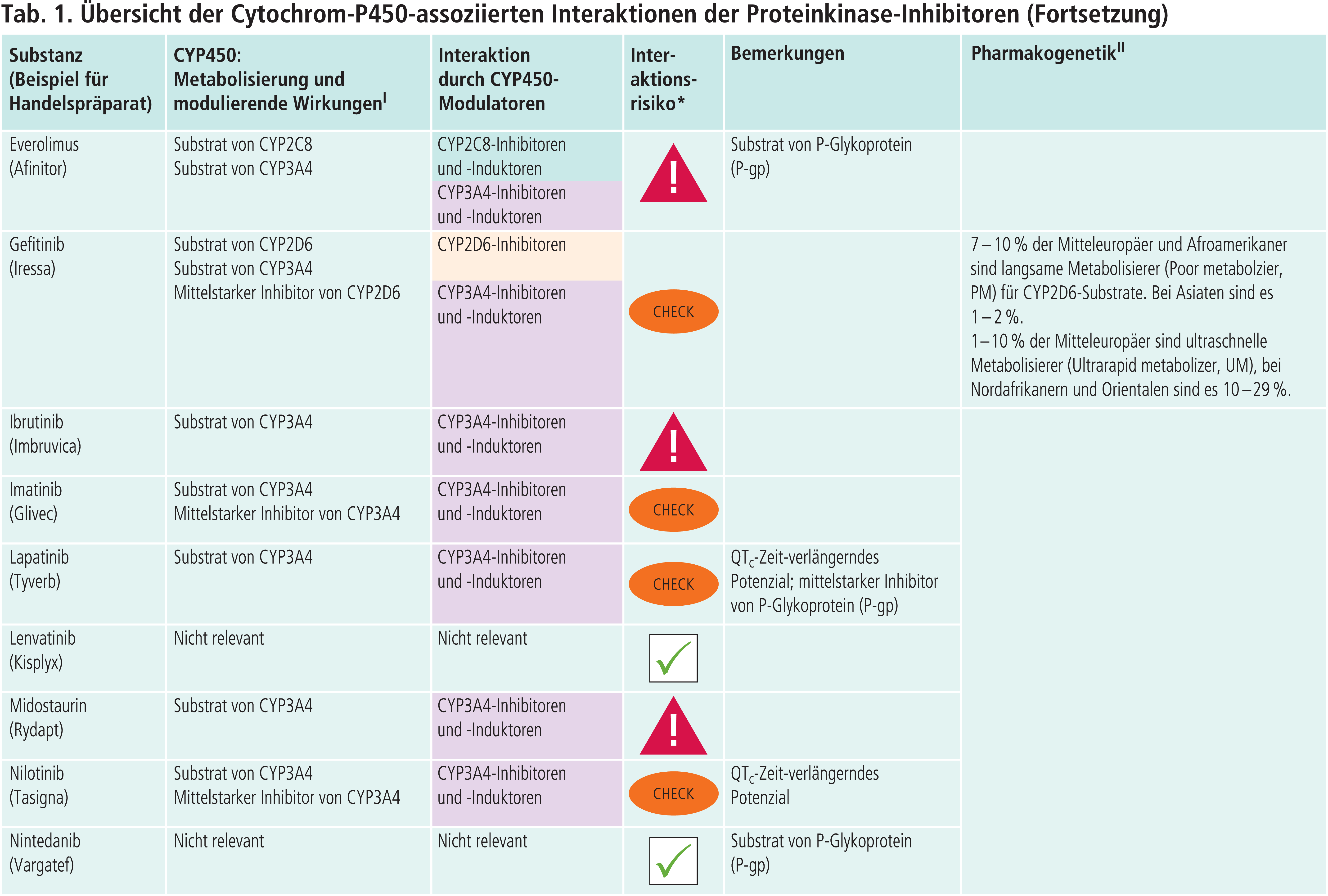
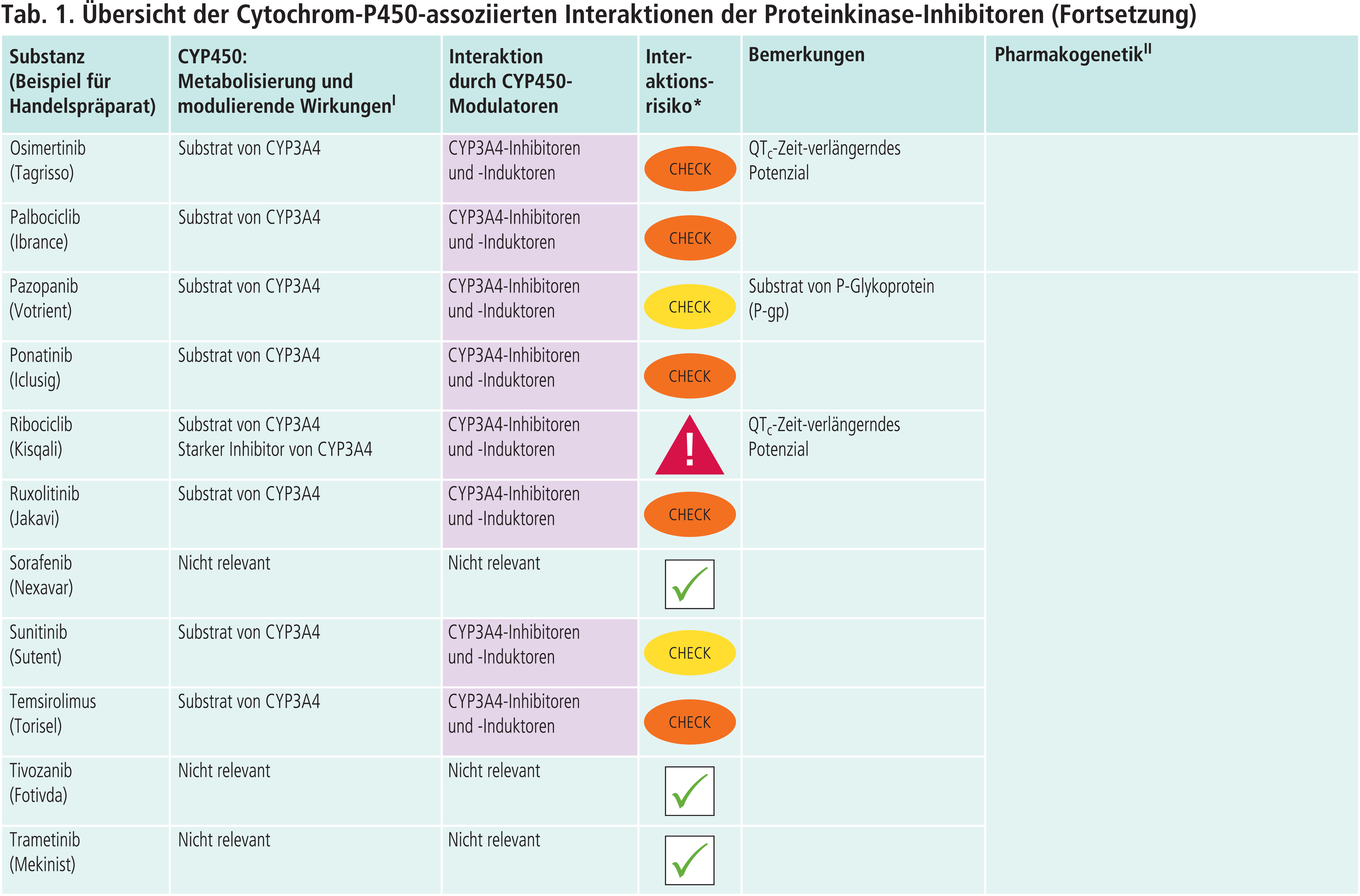
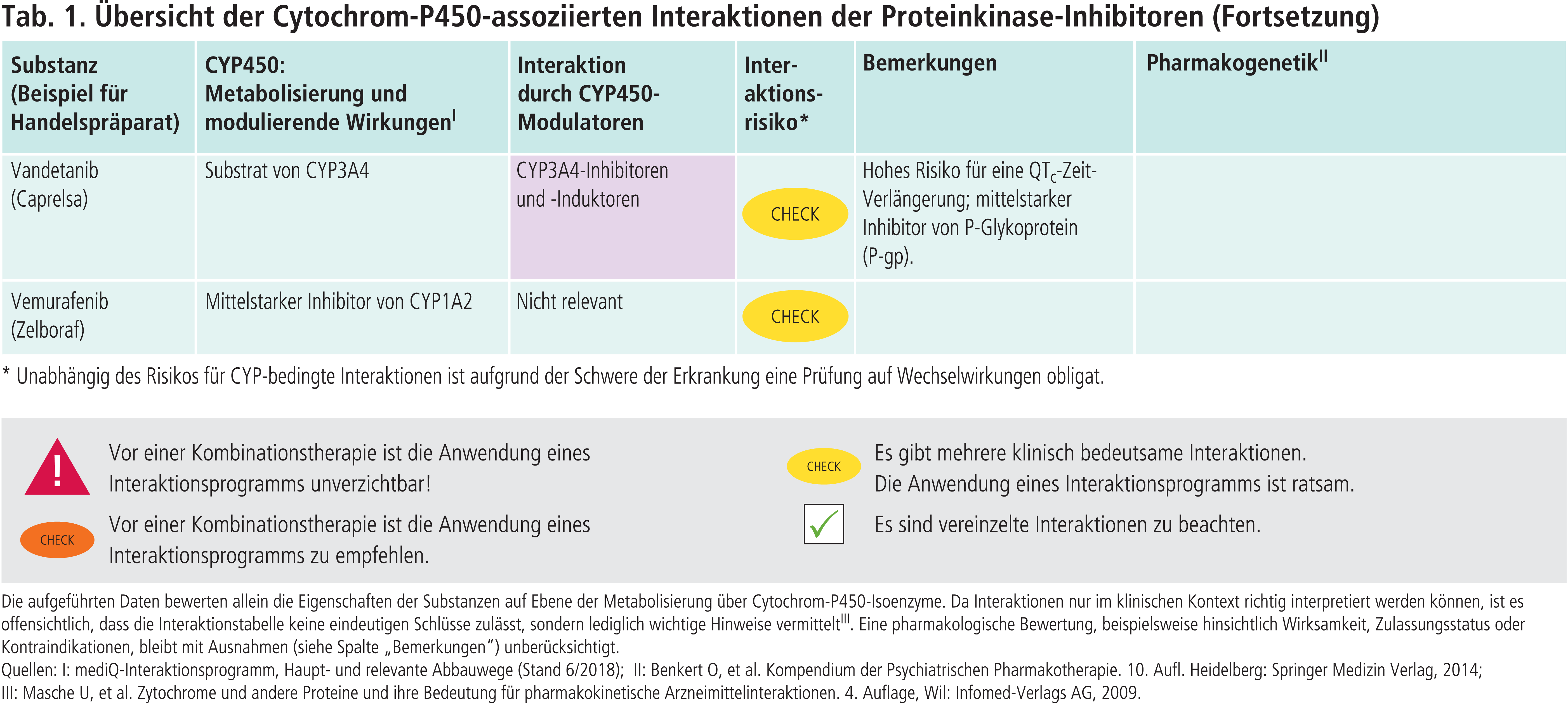
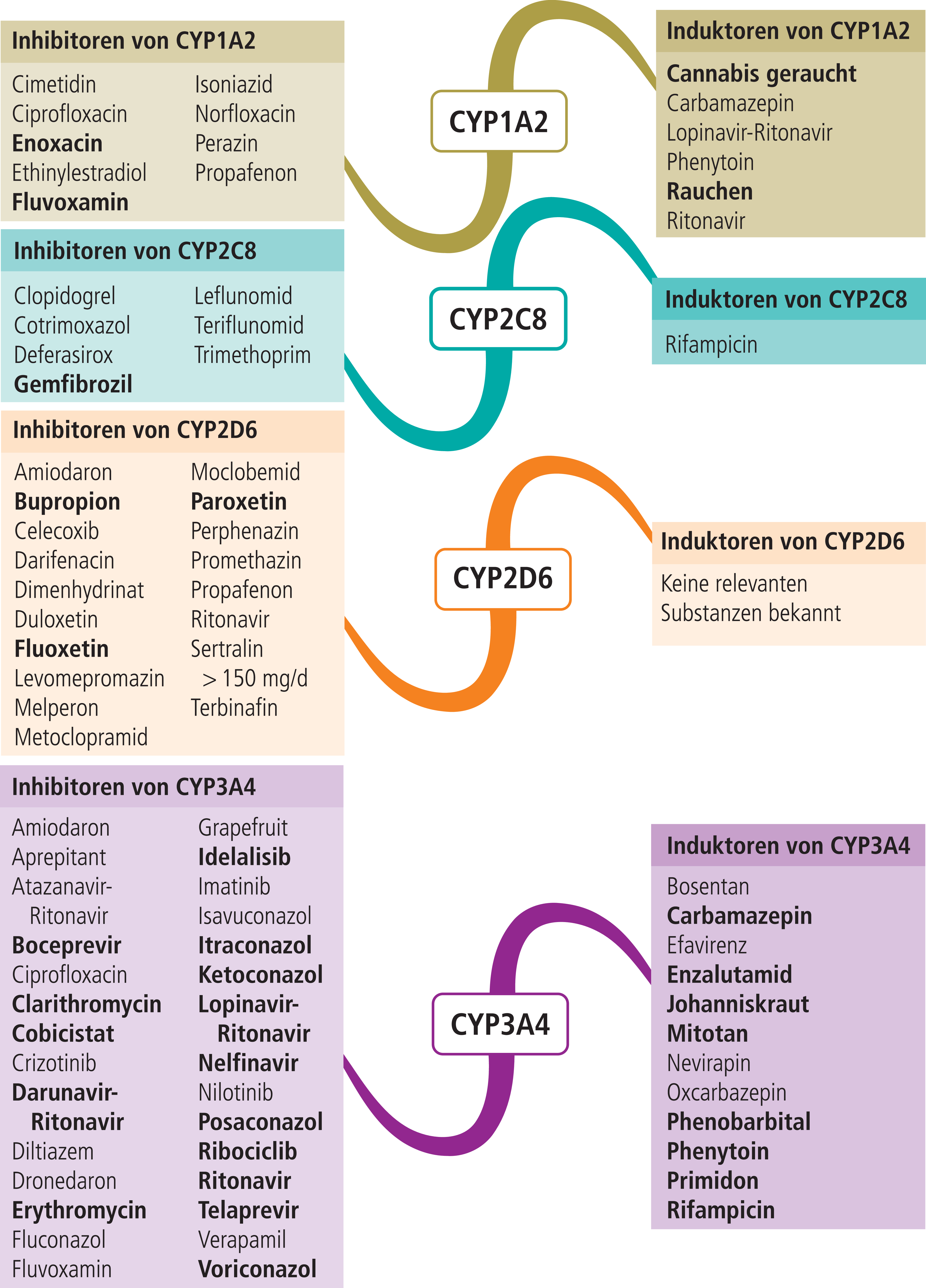
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 1A2, 2C8, 2D6 und 3A4 (Stand 6/2018) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Johnson FM, Agrawal S, Burris H, et al. Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer 2010;116:1582–91.
2. Ouatas T, Duval V, Sinclair K, et al. Concomitant use of midostaurin with strong CYP3A4 inhibitors: an analysis from the RATIFY trial. Poster presentation – 59th Annual Meeting of American Society of Hematology. December 9 – December 12, 2017, Atlanta, GA. Poster 3814.
3. Petri H. Analyse von CYP450-Wechselwirkungen. www. krankenhauspharmazie.de/serien-in-der-kph/analyse-voncyp450-wechselwirkungen.htmlkrankenhauspharmazie.de/serien-in-der-kph/analyse-voncyp450-wechselwirkungen.html (letzter Zugang 22.05.2018).
4. Rote Liste. Fachinfo-Service. https://fachinfo.de/https://fachinfo.de/ (letzter Zugang 29.01.2018).
5. Rowland A, van Dyk M, Mangoni AA, et al. Kinase inhibitor pharmacokinetics: comprehensive summary and roadmap for addressing inter-individual variability in exposure. Expert Opin Drug Metab Toxicol 2017;13: 31–49.
6. Van Erp NP, Gelderblom H, Karlsson MO, et al. Influence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib. Clin Cancer Res 2007;13:7394–400.
7. Wenzel-Seifert K, Ostermeier CP, Omar NB, et al. Unerwünschte kardiovaskuläre Wirkungen von Psychopharmaka. Psychopharmakotherapie 2013;20:148–57.
8. Wille H. Arzneimittelinteraktionen durchGrapefruitsaft.Arzneiverordnung in der Praxis 2014;41:16–9.10.
9. Woosley RL, Romero KA. CredibleMeds. www.crediblemeds.orgwww.crediblemeds.org (letzter Zugang am 22.05.2018).
*Nachdruck aus Krankenhauspharmazie 2018;39:311–8.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2018; 25(04):211-218