Walter E. Müller, Worms/Frankfurt a.M., und Gerd Laux, Soyen/Waldkraiburg/München
Allgemeine Psychopharmakotherapie
Definition, Einteilung und Klassifikation
Psychopharmaka sind Substanzen, die gestörte Stoffwechselprozesse im Gehirn beeinflussen und sie bei Fehlregulationen normalisieren können.
Im weitesten Sinne ist jede in therapeutischer Absicht gegebene Substanz, die in die Steuerungsfunktionen des zentralen Nervensystems eingreift und seelische Abläufe verändert („psychotroper Effekt“), ein Psychopharmakon.
Dieser Begriff ist sehr weit gefasst und beinhaltet beispielsweise auch zentral wirksame Analgetika, Parkinsonmittel und Antiepileptika.
Übliche Einteilung:
- Antidepressiva
- Mood Stabilizer/Stimmungsstabilisierer
- Antipsychotika/Neuroleptika
- Tranquilizer
- Hypnotika
- Antidementiva
- Psychostimulanzien
- Entzugs- und Entwöhnungsmittel
Weitere Bezeichnungen, die sich bei der Klassifikation von Psychopharmaka finden, sind unter anderen Antimanika (Mittel zur Behandlung der Manie) – hierzu zählen Neuroleptika/Antipsychotika und Stimmungsstabilisierer – Sedativa (Beruhigungsmittel), Anxiolytika (Mittel gegen Angsterkrankungen) sowie Antiaddiktiva (Entzugs- und Entwöhnungsmittel).
Enge Beziehungen zu psychischen Erkrankungen besitzen moderne „Lifestyle“-Medikamente wie Sexualtherapeutika (Sildenafil u.a.) sowie Mittel zur Gewichtsreduktion (Orlistat) und Raucherentwöhnung (Bupropion, Vareniclin).
Immer wieder werden neue Klassifizierungen vorgeschlagen, jüngst die an pharmakologischen Wirkungsmechanismen orientierte „Neuroscience-based Nomenclature (NbN)“. Manche Einteilungen orientieren sich an der chemischen Struktur (konnten sich jedoch nicht durchsetzen, da chemisch nahverwandte Stoffe klinisch oft sehr unterschiedliche Wirkungen hervorrufen), andere gehen von den biochemischen oder neurophysiologischen Wirkungsmechanismen aus.
Eine alte Klassifikation, die bereits 1957 von Delay vorgeschlagen wurde, wird zum Teil noch eingesetzt: Sowohl die Weltgesundheitsorganisation (WHO) als auch die europäische Arzneimittelbehörde (EMA) verwenden in der sogenannten ATC-Klassifikation (Anatomisch – Therapeutisch – Chemisch) für die Einteilung der Psychopharmaka die Untergruppen „Psycholeptika“ und „Psychoanaleptika“.
- Psycholeptika: Mittel mit vorwiegend dämpfender Wirkung auf die Psyche – beinhalten Antipsychotika, Tranquilizer/Anxiolytika sowie Hypnotika/Sedativa
- Psychoanaleptika: mit vorwiegend anregender Wirkung auf die Psyche – umfassen Antidepressiva, Stimmungsstabilisierer, Psychostimulanzien und Antidementiva.
Diese Klassifikation ist eigentlich heute vollständig überholt, da Sedierung sich auf eine Beeinflussung der Vigilanz bezieht und nicht notwendig ist für anxiolytische und antipsychotische Wirkung (viele der aktuell eigesetzten Antipsychotika und Anxiolytika sind nicht sedierend). Auf der anderen Seite wirken viele Antidepressiva und Stimmungsstabilisatoren nicht aktivierend.
Fazit
Exakte Abgrenzungen zwischen den einzelnen Psychopharmakagruppen sind nicht immer möglich. Untersuchungen zur Überprüfung der Wirkeigenschaften sowie die teilweise sich überschneidenden Anwendungsgebiete neuerer Substanzen weisen darauf hin, dass die Übergänge zwischen Antipsychotika, Antidepressiva, Stimmungsstabilisierern und Tranquilizern fließend sein können und zum Teil dosisabhängig sind.
Allgemeine Pharmakologie
Pharmakokinetische Grundlagen
Die Wirkung eines Medikaments wird von seiner Konzentration am Wirkort bestimmt. Da die Arzneimittelkonzentration im Gehirn am Menschen nicht messbar ist, muss man sich bei der Betrachtung der pharmakokinetischen Grundbedingungen auf die Wirkstoffkonzentration im Blut verlassen. Diese steht auch in der Regel im Gleichgewicht mit der Konzentration im ZNS. Dies gilt besonders auch für die meisten Psychopharmaka als gut fettlösliche (lipophile) Substanzen, die schnell durch die Blut-Hirn-Schranke penetrieren können.
Typischerweise nimmt der Blutspiegel eines Pharmakons nach oraler Gabe mit der Zeit langsam zu, erreicht bei ausreichender Dosis den therapeutischen Bereich (Invasionsphase), verweilt für eine bestimmte Zeit im therapeutisch benötigten Plasmakonzentrationsbereich und wird dann durch unterschiedliche Eliminationsprozesse langsam ausgeschieden (Evasionsphase) (Abb. 1). Bei akuter Anwendung kommt es meist auf einen möglichst schnellen Wirkungseintritt an, das heißt das möglichst schnelle Erreichen einer ausreichenden Konzentration der Substanz am Wirkort (ZNS). Die Geschwindigkeit dieses Vorgangs lässt sich aus den sogenannten tmax-Angaben abschätzen, der Zeit, die eine Substanz benötigt, bis maximale Plasmaspiegel erreicht sind.
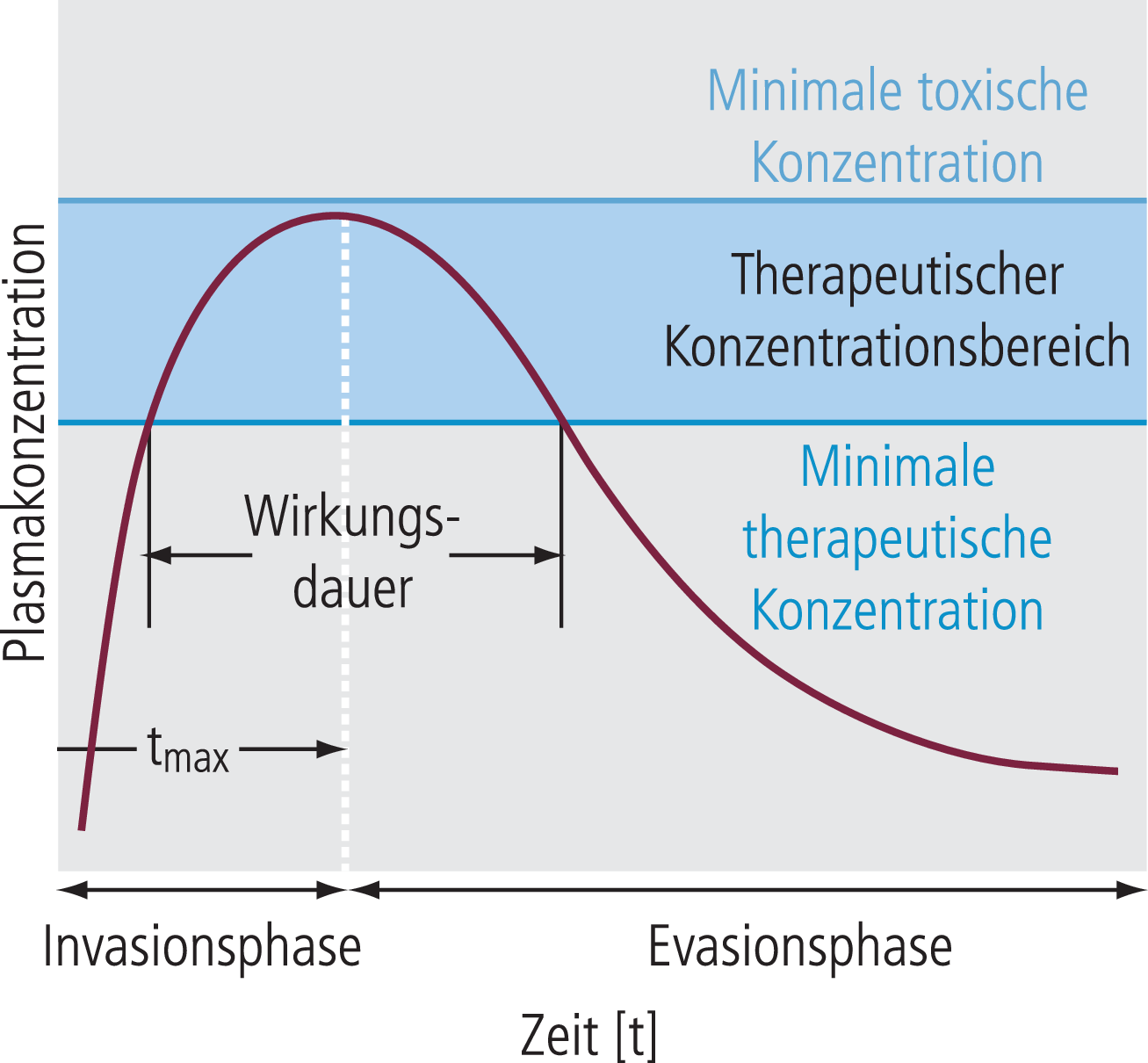
Abb. 1. Schematische Darstellung eines Plasmaspiegelverlaufs nach oraler Applikation
Ziel ist immer, den therapeutischen Konzentrationsbereich zwischen minimaler therapeutischer und minimaler toxischer Konzentration zu erreichen bzw. den Bereich mit nicht mehr akzeptablen unerwünschten Arzneimittelwirkungen zu vermeiden.
Für die Zeit bis zum Erreichen von tmax spielt bei festen oralen Arzneiformen (Tabletten, Kapseln, Dragees) die Zerfallszeit und die Auflösungszeit eine wesentliche Rolle, das heißt die Zeit, die vergeht, bis nach Einnahme des Medikaments ausreichend gelöste Moleküle im Magen- bzw. Darminhalt zur Resorption zur Verfügung stehen (etwa 30 bis 60 Minuten). Diese Verzögerung kann für die Anwendung in der Akutsituation bei einigen Substanzen durch Einnahme von flüssigen oralen Arzneiformen (hauptsächlich Tropfen), aber auch durch andere spezielle galenische Zubereitungen wie lyophilisierte Plättchen (z.B. Lorazepam, Tavor Expidet®) oder sublinguale Tabletten (Asenapin, Sycrest®) umgangen werden, womit nach oraler Einnahme sehr schnell ausreichende Wirkspiegel im Gehirn erreicht werden können (innerhalb von Minuten). Ein weiteres Beispiel für eine schnelle nicht parenterale Applikation ist die inhalative Arzneiform (z.B. für das Antipsychotikum Loxapin; Adasuve®) bei akuter Agitation.
In Fällen einer akuten medikamentösen Intervention, wenn eine orale Medikation nicht (mehr) möglich ist, wird eine parenterale Applikation unumgänglich. Hier erreicht man praktisch zum Zeitpunkt der Bolusinjektion schon maximale Blutspiegel (Abb. 2), die bei ausreichend lipophilen Substanzen sehr schnell zu hohen Konzentrationen im ZNS führen. Andere parenterale Applikationsformen wie beispielsweise die intramuskuläre Injektion können bei einzelnen Substanzen auch infrage kommen, führen aber in der Regel wesentlich langsamer zu ausreichenden ZNS-Konzentrationen.
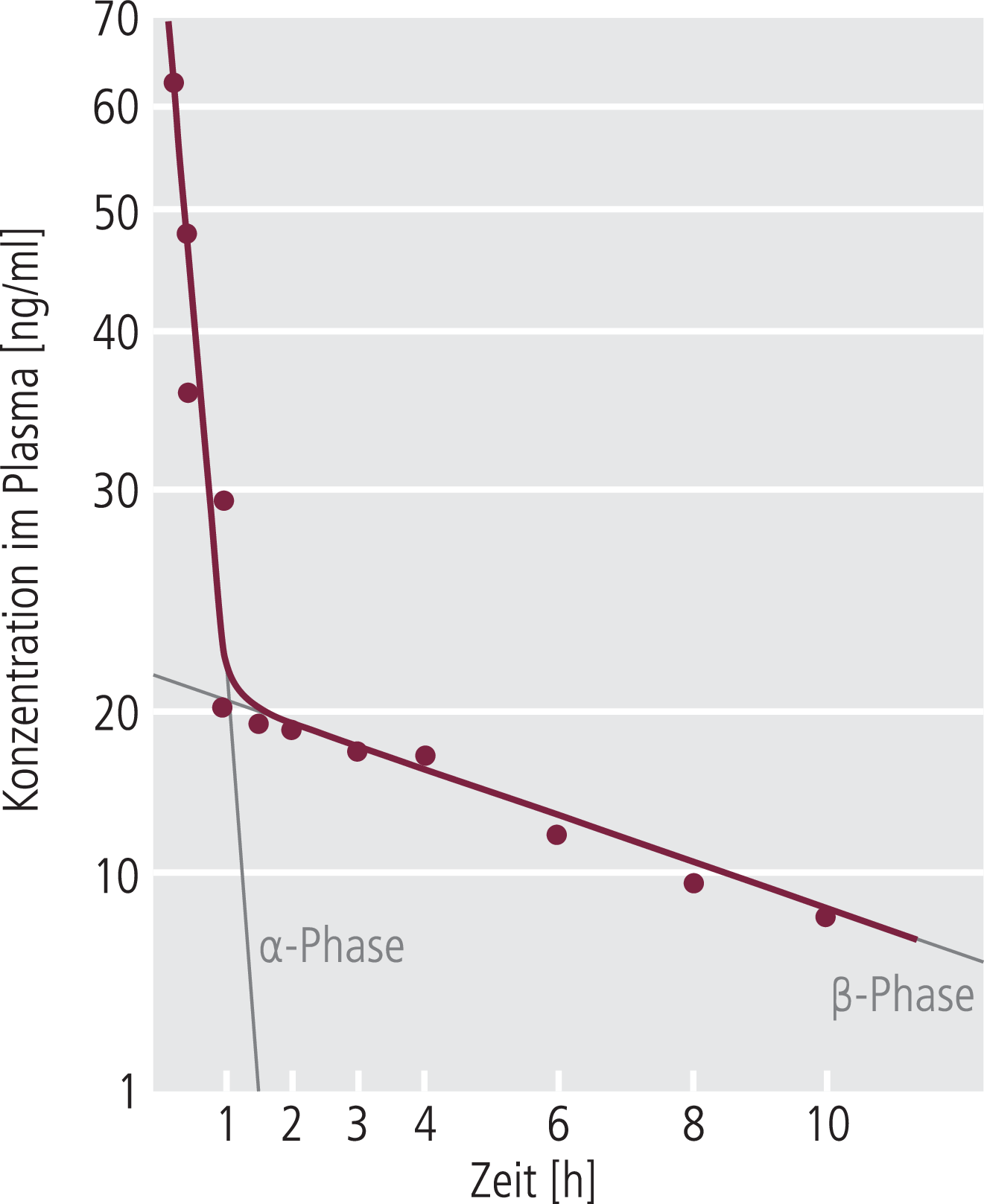
Abb. 2. Plasmaspiegelverlauf nach i.v. Applikation in halblogarithmischer Auftragung (am Beispiel i.v. Injektion von 10 mg Verapamil; nach Hamann et al.,Clin Pharmacokin 1984;9:26). Die Plasmaspiegelverlaufskurve kann in zwei lineare Phasen aufgeteilt werden: die α-Phase, bei der die Abnahme des Plasmaspiegels durch Verteilung ins Gewebe bestimmt ist, und die β-Phase, die die terminale Elimination beschreibt. Die Zeit, in der der Plasmaspiegel in der β-Phase um die Hälfte abnimmt, wird als Eliminationshalbwertszeit (t1/2) bezeichnet. Sie bezieht sich auf die Abnahme des Plasmaspiegels, der meistens mit der Konzentration in den Geweben im Gleichgewicht steht (Ausnahme ist das längere Verweilen von einigen Psychopharmaka im ZNS relativ zum Plasmaspiegel).
Die therapeutische Wirkung eines Arzneistoffs wird generell durch das Unterschreiten des minimalen therapeutischen Plasmaspiegels terminiert (in der Evasionsphase; Abb. 1). Bei vielen Substanzen kann dieser Evasionsprozess in zwei Phasen aufgeteilt werden, eine sogenannte α-Phase mit einer kurzen und eine β-Phase mit einer längeren Zeitkonstanten (Abb. 2). Die α-Phase, die im angegebenen Beispiel sehr deutlich ausgeprägt ist, wird in der Regel von Umverteilungsphänomenen getragen. Nach akuter (auch oraler) Gabe erreicht ein lipophiler Wirkstoff in kürzester Zeit (Minuten) sehr hohe Konzentrationen im ZNS, da dieses Organ besonders gut durchblutet wird. Entsprechend seinem bei lipophilen Substanzen hohen Verteilungsvolumen kommt es aber dann im Anschluss zu einer relativ schnellen Umverteilung aus dem gut durchbluteten Organ ZNS in weniger gut durchblutete Bereiche wie beispielsweise die Muskelmasse. Bei ausreichend fettlöslichen Substanzen kann allein diese Umverteilungsphase bedeuten, dass die minimale therapeutische Konzentration im ZNS unterschritten und die akute Wirkung im ZNS nur durch diese Umverteilungsphänomene terminiert wird. Dieses Phänomen gilt besonders bei akuten Einmaldosierungen, für welche die kurzen Wirkdauern häufig nicht mit längeren Eliminationshalbwertszeiten übereinstimmen (s. β-Phase in Abb. 2).
Bei den meist lipophilen Psychopharmaka sind häufig die Gewebekonzentrationen deutlich höher als die Plasmakonzentrationen. Solche Substanzen haben ein großes Verteilungsvolumen (VD; Maß für die Verteilung eines Arzneistoffs im Körper: Quotient aus der Gesamtmenge des verabreichten Arzneistoffs und seiner Blut- bzw. Plasmakonzentration) und meistens schlechte Korrelationen zwischen Plasmakonzentration und Wirkung.
Im typischen Plasmaspiegelverlauf (Abb. 1) überwiegt am Anfang in der Invasionsphase die Resorption aus dem Magen-Darm-Trakt. Der im Blut vorhandene Arzneistoff erreicht allerdings schon in dieser Phase die Eliminationsorgane (Niere, Magen-Darm-Trakt, Lunge usw.). Ist die Resorption abgeschlossen, überwiegt in der Evasionsphase die Elimination. Unter Elimination versteht man die Summe der Mechanismen, die zum Verschwinden des Arzneistoffs aus dem Körper führen, wie sie von der terminalen Plasmakonzentrationskurve beschrieben werden (Abb. 1 und 2). Dies darf nicht verwechselt werden mit der Eliminationshalbwertszeit oder der Beschreibung der Elimination über den Plasmaspiegel; hier bezieht man sich nur auf die Abnahme im Blut, was wie erwähnt nicht mit einer identischen Abnahme des Arzneistoffs im gesamten Organismus parallel gehen muss.
Elimination kann direkt sein (der Wirkstoff verlässt direkt den Körper, z.B. über die Niere, die Fäzes oder die Lunge). Direkte Elimination über die Niere spielt allerdings bei Psychopharmaka nur eine geringe Rolle, da die meisten der in der Regel lipophilen Arzneistoffe zwar in der Niere filtriert werden, dann aber über passive Diffusion im Tubulus-System wieder rückresorbiert werden. Sie müssen erst über die komplexen Mechanismen der Biotransformation so lange chemisch verändert werden (metabolische Elimination), bis sie wasserlöslich genug sind, um dann über die Nieren ausgeschieden zu werden. Ziel der Biotransformation ist nicht der Wirkverlust (Entgiftung), sondern die Erhöhung der Wasserlöslichkeit (Abb. 3).
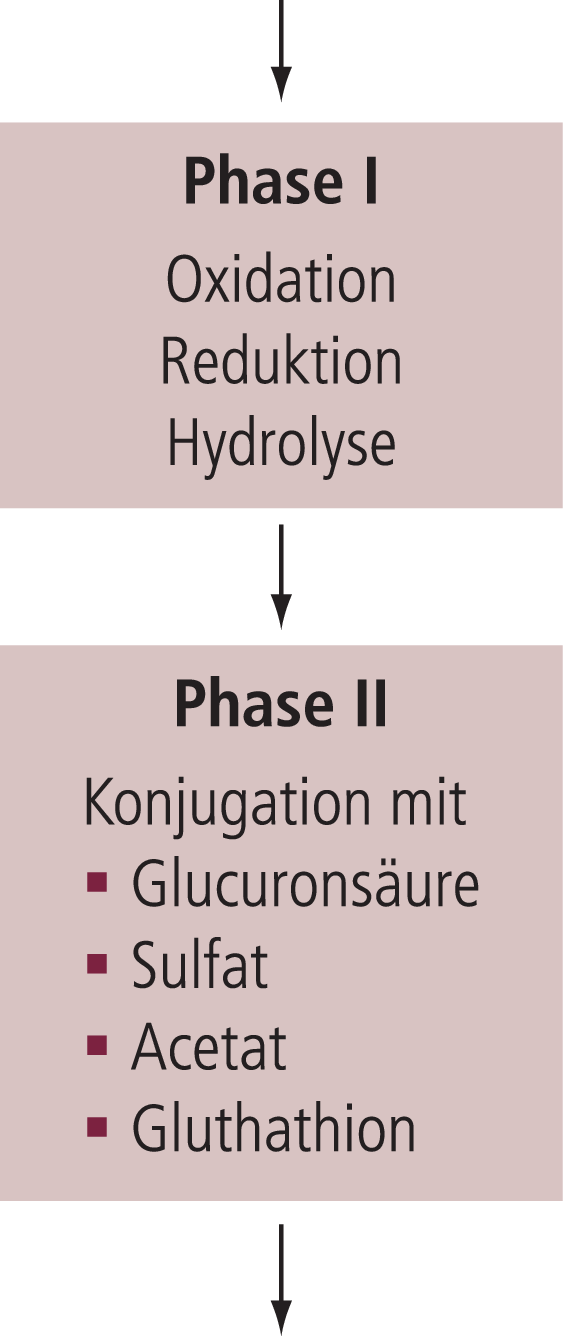
Abb. 3. Biotransformation von Wirkstoffen (Arzneistoffmetabolismus). Zur terminalen Ausscheidung über die Niere müssen die ursprünglichen Wirkstoffmoleküle ausreichend wasserlöslich (hydrophil) gemacht werden. Dies geschieht über die komplexen enzymatischen Mechanismen der Biotransformation. In vielen Fällen muss erst eine reaktive Gruppe chemisch eingeführt werden, die dann die Ankoppelung eines hydrophilen Moleküls erlaubt, womit erst der wasserlösliche Metabolit entsteht. Nur in Einzelfällen ist eine Phase-II-Reaktion ausreichend.
Die Biotransformation von Wirkstoffen (Arzneistoffmetabolismus) wird grob in zwei Schritte eingeteilt. In der Phase I muss der Wirkstoff chemisch modifiziert werden (z.B. Oxidation, Reduktion, Hydroxylierung, Esterhydrolyse), um entweder direkt oder erst nach Konjugation in einer sich anschließenden Phase-II-Reaktion renal eliminiert werden zu können. In der Phase II wird der Wirkstoff durch die Konjugation mit einem hydrophilen Substrat (z.B. Glucuronsäure, Sulfat) ausreichend wasserlöslich für die renale Elimination. Die dafür notwendige reaktive Gruppe wird häufig erst in der Phase I eingeführt.
Ist das therapeutische Ziel der Erhalt einer ausreichenden Plasmakonzentration über längere Zeit (Tage oder Wochen), wird man dies durch Mehrfacheinnahme erreichen, wobei die Höhe der angestrebten mittleren Plasmakonzentration von der individuellen Dosis, dem Dosierungsintervall und vor allen Dingen auch der Eliminationshalbwertszeit der jeweiligen Substanz bestimmt wird. Bei kurzer Halbwertszeit (wenige Stunden) muss dabei die Substanz mehrfach tagsüber eingenommen werden, um den Plasmaspiegel im Wirkbereich zu halten (Abb. 4). Bei konstanter multipler Dosierung wird in der Regel nach etwa fünf bis sieben Eliminationshalbwertszeiten der Steady-State (Fließgleichgewicht) erreicht, also die Phase, bei der ein mittlerer Plasmaspiegel konstant gehalten wird, da sich dann die Invasions- und der Evasionsprozesse die Waage halten (die Summe dieser beiden Prozesse bleibt in etwa konstant). Der gleiche Zeitraum gilt für jede Dosisänderung. Hat die Substanz eine kurze Halbwertszeit, sind die Plasmaspiegel schon nach wenigen Stunden unter der minimalen Wirkkonzentration, sodass eine Mehrfacheinnahme über den Tag notwendig wird. Depotarzneiformen (retardierte Arzneiformen) setzen den Wirkstoff bedingt durch verschiedene Techniken über viele Stunden frei, sodass man hier mit einer Einzelgabe über den Tag auskommt (Abb. 4).
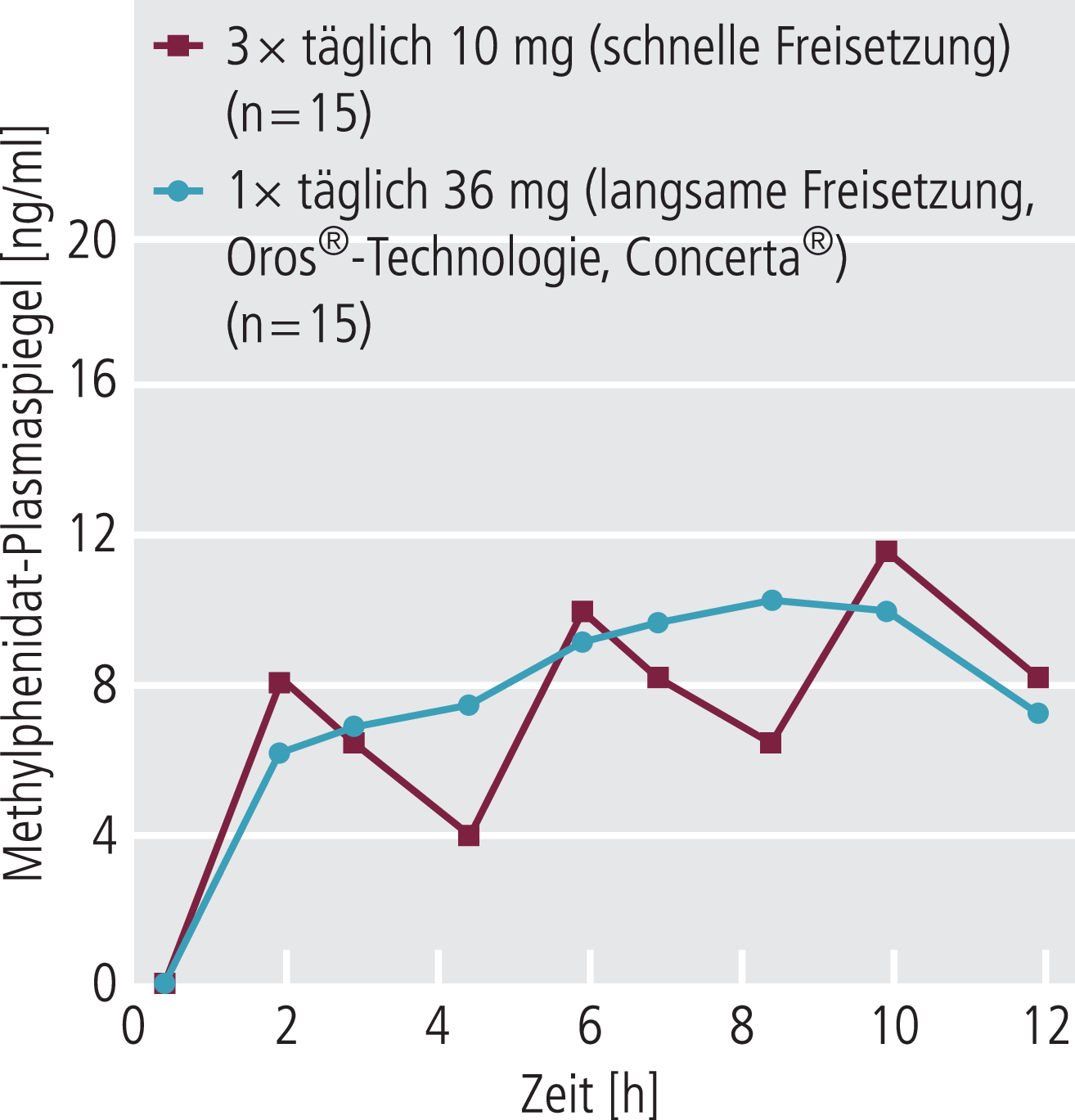
Abb. 4. Methylphenidat-Plasmaspiegel nach mehrfacher Einfachdosierung (dreimal täglich) einer rasch freisetzenden und nach Einnahme (einmal am Morgen) einer retardierten Arzneiform (nach Volkow et al. Am J Psychiatry 2003;160:1909–18). Um den Plasmaspiegel tagsüber im therapeutischen Bereich zu halten, muss die normale orale Arzneiform dreimal im Abstand von einigen Stunden eingenommen werden. Dies kann bei der retardierten oralen Arzneiform (langsame Freigabe des Wirkstoffs im Magen-Darm-Trakt über Stunden) mit einer Einzelgabe am Morgen erreicht werden.
Allgemeines zum Wirkungsmechanismus (Pharmakodynamik)
Die chemische Neurotransmission als der primäre Angriffspunkt
Alle wichtigen Funktionen des zentralen Nervensystems (ZNS) wie Aufnahme, Verarbeitung und Speicherung sensorischer Informationen, aber auch psychische wie motorische Reaktionen auf entsprechende sensorische Informationen werden über Funktionsänderungen von Nervenzellen vermittelt. Um dieser Vielfalt an komplexen Aufgaben gerecht zu werden, ist für eine optimale Funktion des ZNS eine intensive Kommunikation zwischen den einzelnen Nervenzellen essenziell. Diese wird im ZNS größtenteils über chemische Neurotransmission vermittelt. Hierbei sind Zellkörper, Dendriten und Axone durch eine große Anzahl von Synapsen miteinander verschaltet.
Die funktionelle Vielfalt biologischer Prozesse des ZNS spiegelt sich deshalb im Wesentlichen auf synaptischer Ebene wider, wo eine Fülle unterschiedlicher Neurotransmittersubstanzen nach Freisetzung aus dem präsynaptischen Neuron mit Rezeptoren des postsynaptischen Neurons interagiert; das aufgenommene Signal wird dann über verschiedene Transduktionsmechanismen in das rezeptive Neuron weitergegeben (Abb. 5). Bezogen auf die große Zahl von Nervenzellen im ZNS (ca. 50 Milliarden) ist die Zahl der zur Verfügung stehenden Neurotransmitter sehr gering, sodass die große Breite von Funktionsabläufen nicht auf eine Transmitterselektivität, sondern auf die Beteiligung unterschiedlicher Nervenzellen bzw. unterschiedlicher Synapsen an einer Nervenzelle zurückgeht. Sehr vereinfacht bedeutet dies, dass praktisch jeder Transmitter mehr oder weniger an allen Funktionsabläufen beteiligt ist. Dies ist noch nachvollziehbar bei GABA oder L-Glutamat, deren Synapsen je etwa ein Drittel aller Synapsen im ZNS ausmachen. Dies gilt aber auch für die modulierenden Amin-Neurotransmitter wie Serotonin, Dopamin und Noradrenalin, die bezogen auf die Gesamtzahl in nur einer extrem kleinen Anzahl von Nervenzellen vorkommen; zum Beispiel gibt es weniger als 100000 noradrenerge Neurone. Die Selektivität entsteht dadurch, dass der freigesetzte Transmitter in der Regel nur an der Synapse wirkt, wo er freigesetzt wird und wo er in Millisekunden inaktiviert wird.
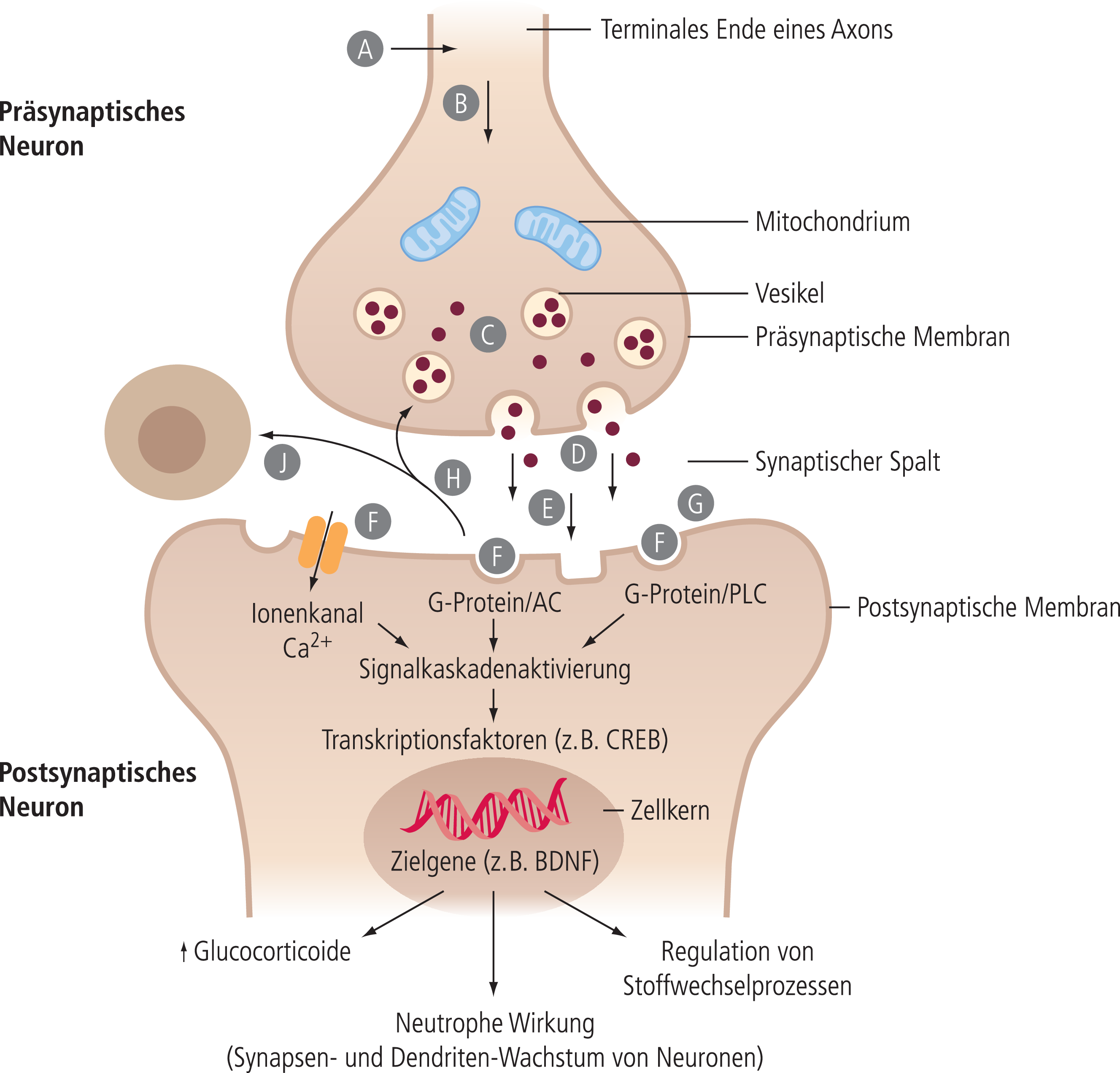
Abb. 5. Schematische Darstellung einer Synapse als Kommunikationsprinzip zwischen zwei Nervenzellen. Der Transmitter selbst – oder meist seine Vorstufe – wird von spezifischen Systemen ins Neuron aufgenommen (A). Der aufgenommene bzw. aus der Vorstufe im Neuron synthetisierte Transmitter wird über den axonalen Transport an die Nervenendigungen transportiert (B) und dort in Vesikeln gespeichert (C). Infolge eines Aktionspotenzials des Axons und eines damit verbundenen Ca2+-Einstroms wird der Transmitter durch Exozytose aus den Vesikeln in den synaptischen Spalt freigesetzt (D) und kann nach Diffusion (E) mit Rezeptoren auf der postsynaptischen Seite reagieren (F). Die Inaktivierung des Transmitters erfolgt durch Abbau und/oder Aufnahme an der postsynaptischen Seite (G), durch Rückdiffusion (H) und Aufnahme über spezifische Transporter ins präsynaptische Neuron bzw. in Synapse-begleitende Gliazellen (J). Das von den Rezeptoren für den Neurotransmitter auf der postsynaptischen Seite aufgenommene Signal wird über unterschiedliche zum Teil G-Protein-gekoppelte Transduktionssysteme in die Zelle geleitet (z.B. Rezeptor-gekoppelte Ionenkanäle, Adenylatcyclase, Phospholipase C). Die dann folgende Aktivierung komplexer intrazellulärer Signalkaskaden wird initial über die Aktivierung sekundärer Transmitter wie cAMP, IP3 oder auch Ca2+-Ionen vermittelt. Die Signalkaskaden führen dann letztlich zu Veränderung des Membranpotenzials (Hyperpolarisation oder Depolarisation) mit der Konsequenz einer Hemmung bzw. Initiierung eines Aktionspotenzials oder zur Freisetzung von Transkriptionsfaktoren, die nach Diffusion in den Zellkern die Synthese von Signalproteinen (z.B. des neurotrophen Faktors BDNF) initiieren.
Die Informationsweitergabe wird auf der postsynaptischen Seite von Rezeptoren übernommen, die vom freigesetzten Transmitter besetzt werden und die das so ausgelöste Signal dann über verschiedene Transduktionsmechanismen in das Neuron weiterleiten. Transduktionsmechanismen können im einfachsten Fall Ionenkanäle sein, zum Beispiel ein Chloridkanal, der durch den Neurotransmitter Gamma-Aminobuttersäure (GABA) geöffnet wird. Die meisten Transduktionsmechanismen sind aber membranständige Enzyme, die an den eigentlichen Rezeptor über ein stimulierendes oder inhibitorisches G-Protein gekoppelt sind. Ihre Aktivierung führt zur intrazellulären Bildung oder Freisetzung sekundärer Transmitter (zyklisches AMP [cAMP], Inositolphosphate, Calcium-Ionen), die ihrerseits die Funktionsänderung der Nervenzelle auslösen. Hier spielt dann die Aktivierung von Transkriptionsfaktoren für zellulare Signalmoleküle (z.B. neurotrophe Faktoren) eine wichtige Rolle.
Die heute therapeutisch zur Verfügung stehenden Psychopharmaka greifen alle mehr oder weniger spezifisch in einzelne Prozesse der chemischen Neurotransmission bestimmter Neurotransmittersysteme ein. Im Vordergrund stehen hier direkte Effekte an den Rezeptoren (Agonisten, Antagonisten) oder Effekte auf die jeweiligen Inaktivierungsmechanismen (Transporter, inaktivierende Enzyme). Ihre Wirkung ist nur dann spezifisch für das ZNS, wenn das jeweilige Transmittersystem in der Peripherie keine Rolle spielt (z.B. bei den Benzodiazepinen als GABAerge Substanzen, bei Memantin als antiglutamaterge Substanz, oder auch die Antipsychotika als antidopaminerge Substanzen, da Dopamin in der Peripherie fast keine Rolle spielt). Im Gegensatz zu den synaptisch freigesetzten und sofort wieder inaktivierten Neurotransmittern ist die Wirkung der Psychopharmaka funktionell unspezifisch, da sie per Diffusion alle Bereiche des ZNS eher gleichmäßig erreichen und überall dort wirken, wo das pharmakologische Target (Rezeptor, Transporter, Enzym) vorhanden ist.
Klinische Psychopharmakologie: Antidepressiva
Pharmakologische Wirkung
Für die Therapie stehen heute über 20 unterschiedliche Antidepressiva zur Verfügung, die über unterschiedliche Angriffspunkte (Targets) in die chemische Neurotransmission des Gehirns eingreifen.
- Diese akuten Angriffspunkte der Antidepressiva sind bis auf agonistische Effekte an Melatonin-Rezeptoren alle mit einer Veränderung der Neurotransmission in den zentralen noradrenergen, serotoninergen und dopaminergen Systemen verbundeBlockade der Rücktransporter von Noradrenalin bzw. Serotonin (5-HT) bzw. Dopamin (Wiederaufnahmehemmung),
- Blockade präsynaptischer Autorezeptoren (Alpha 2),
- Hemmung des Abbaus biogener Amine (MAO-Hemmung),
- 5-HT2-Antagonismus,
- Melatonin-MT1- und MT2-Agonismus.
Diese akuten Angriffspunkte sind auch die Basis der aktuellen Klassifikation der Antidepressiva (Tab. 1). Der ursprüngliche Begriff trizyklische Antidepressiva (basierend auf der chemischen Grundstruktur) wird heute abgelöst durch die Bezeichnung nichtselektive Monoamin-Wiederaufnahmehemmer. Die zu den Trizyklika gezählten Substanzen Trimipramin und Opipramol, das heute eher als Anxiolytikum gesehen wird, sind allerdings keine Monoamin-Wiederaufnahmehemmer und passen in der neueren Klassifizierung am besten zu den Rezeptorantagonisten.
Tab. 1. Pharmakologische Einteilung von Antidepressiva
|
Substanzgruppen |
Wirkstoffe (Handelsnamen an Beispielen) |
|
|
Nichtselektive Monoamin-Wiederaufnahmehemmer (NSMRI) |
Trizyklische Antidepressiva (TZA) |
z.B. Amitriptylin (Saroten), Doxepin (Aponal), Clomipramin (Anafranil) |
|
Selektive Monoamin-Wiederaufnahmehemmer |
Serotonin-Wiederaufnahmehemmer (SSRI) |
z.B. Escitalopram (Cipralex), Fluoxetin (Fluctin), Sertralin (Zoloft) |
|
Noradrenalin-Wiederaufnahmehemmer (NARI) |
Reboxetin (Edronax) |
|
|
Duale Wiederaufnahmehemmer |
SNRI: Venlafaxin (Trevilor), Duloxetin (Cymbalta), Milnacipran (Milnaneurax) |
|
|
NDRI: Bupropion (Elontril) |
||
|
Rezeptorantagonisten |
(NaSSA): Mirtazapin (Remergil) |
|
|
Mianserin, Trazodon Trimipramin, Opipramol(?) |
||
|
Monoaminoxidase-Hemmer (MAOH) |
Tranylcypromin (Jatrosom), Moclobemid (Aurorix) |
|
|
Melatoninagonist/Serotoninantagonist |
Agomelatin (Valdoxan) |
|
NaSSA: Noradrenalin-Serotonin selektives Antidepressivum; NDRI: selektiver Noradrenalin-Dopamin-Rückaufnahme-Inhibitor; SNRI: selektiver Serotonin-Noradrenalin-Rückaufnahme-Inhibitor
Nichtselektive, sogenannte trizyklische Antidepressiva (NSMRI, TZA) beeinflussen verschiedene Neurotransmittersysteme über eine Hemmung der Wiederaufnahmesysteme bzw. Hemmung verschiedener Rezeptoren; primärer Effekt für ihre therapeutische Wirkung ist die Hemmung der Wiederaufnahme von Noradrenalin und Serotonin. Die zusätzliche Blockade verschiedener postsynaptischer Rezeptoren (z.B. Acetylcholin oder Histamin) führt zu weiteren Wirkeffekten und auch unerwünschten Arzneimittelwirkungen. (H1-Antagonismus und Sedierung; M1-Antagonismus und anticholinerge Effekte). Serotonin-selektive Antidepressiva (SSRI) bzw. Noradrenalin-selektive Substanzen (NARI) bewirken durch Blockade des präsynaptischen Serotonin- bzw. Noradrenalintransporters eine selektive Hemmung der Wiederaufnahme von Serotonin bzw. Noradrenalin. Infolgedessen kommt es zu erhöhten Konzentrationen dieser Transmitter im synaptischen Spalt. Sogenannte duale Substanzen (Venlafaxin, Duloxetin, Milnacipran) interagieren mit beiden Transporter-Proteinen (SNRI). Auch die Blockade des Transporters für Dopamin (NDRI), wie bei Bupropion, wird für antidepressive Effekte verantwortlich gemacht.
MAO-Hemmer erhöhen die Konzentration von Noradrenalin und Serotonin, indem sie das abbauende Enzym Monoaminoxidase (v.a. MAO-A) hemmen.
Ein weiteres Wirkprinzip ist die Hemmung präsynaptischer Alpha-2-(α2-)Autorezeptoren an der noradrenergen Synapse (Mirtazapin). Die zumindest teilweise Ausschaltung dieses physiologischen Bremsmechanismus, der die Freisetzung von Neurotransmittern reduziert, führt gleichfalls zu einer Erhöhung der synaptischen Verfügbarkeit der beiden Neurotransmitter Serotonin und Noradrenalin.
Agomelatin ist ein Agonist an Melatoninrezeptoren (M1 und M2) und ein Antogonist an 5-HT2C Rezeptoren. Eine Sonderstellung nimmt Tianeptin ein: Es verstärkt die Serotonin-Wiederaufnahme und senkt so extrazelluläre Serotonin-Konzentrationen, es moduliert außerdem direkt die glutamaterge Neurotransmission. Als sogenanntes multimodales Antidepressivum wirkt Vortioxetin antagonistisch auf 5-HT3-, 5-HT1D- und 5-HT7-Rezeptoren, partiell agonistisch auf 5-HT1B-Rezeptoren, agonistisch am 5-HT1A-Rezeptor und ist außerdem ein Serotonin-Wiederaufnahmehemmer. Die Substanz wurde vom Hersteller in Deutschland nach gescheiterten Preisverhandlungen vom Markt genommen.
Trimipramin, das zu den Trizyklika gezählt wird, bewirkt keine Monoamin-Wiederaufnahmehemmung, sondern wirkt antagonistisch an Histamin-, Acetylcholin-, Dopamin-, 5-HT2- und anderen Rezeptoren. Trazodon wirkt an Serotonin-Rezeptoren und am Serotonin-Transporter, zusätzlich an H1- und adrenergen Rezeptoren.
Es konnte gezeigt werden, dass für die Wirkung des Johanniskrautextrakts der Anteil an Hyperforin entscheidend ist, das eine Wiederaufnahmehemmung von Serotonin, Noradrenalin, Dopamin, GABA und L-Glutamat bewirkt, aber auch direkte neurotrophe Eigenschaften über eine direkte Aktivierung eines bestimmten Ca2+-Kanals zeigt.
Praktisch alle akuten Angriffspunkte der Antidepressiva (Tab. 1) liegen in den synaptischen Mechanismen der zentralen serotoninergen, noradrenergen und dopaminergen Neurotransmission, Systemen mit einer jeweils sehr kleinen Anzahl von Nervenzellen, die aber trotzdem praktisch alle Funktionen unseres Organismus im Gehirn modulieren. Störungen in diesen Strukturen könnten daher leicht mit der großen Breite des depressiven Syndroms von psychischen über psychomotorischen zu somatischen Symptomen verbunden sein, die ja auch alle auf die Therapie mit einem Antidepressivum ansprechen. Die damit verbundene Vorstellung der Korrektur einer spezifischen Störung in diesen Systemen durch die Antidepressiva (z.B. serotoninerge-noradrenerge Imbalance-Hypothese) konnte allerdings nie bestätigt werden. Man geht daher heute davon aus, dass diese akuten Effekte nicht direkt mit der antidepressiven Wirksamkeit im Zusammenhang stehen, zumal sie sehr schnell auftreten, die antidepressive Wirksamkeit sich aber langsam über ein bis drei Wochen entwickelt. Adaptive Veränderungen der Neurotransmission (Neuroplastizität), die im ähnlichen Zeitfenster ablaufen, werden daher heute mit der eigentlichen Wirksamkeit der Antidepressiva in Verbindung gesehen (Abb. 6). Hierzu zählen nicht nur adaptive Veränderungen der eigentlichen Neurotransmission (z.B. die Downregulation von Betarezeptoren), sondern besonders auch adaptive Veränderungen an den neuronalen Strukturen (strukturelle Neuroplastizität). Diese betreffen die postsynaptischen Feinstrukturen (Spine-Struktur und Spine-Dichte), aber auch Veränderungen von Dendritenlänge und Dendritenverzweigung und die in wenigen hippocampalen Strukturen mögliche Neusynthese von Nervenzellen (Neurogenese) (Abb. 6) und auf präsynaptischer Seite das Phänomen der Langzeitpotenzierung. Neuroplastizität ist ein wesentlicher Aspekt der Anpassungsfähigkeit unseres Gehirns und scheint bei chronischem Stress und auch Depression gestört zu sein. Eine Normalisierung gestörter Neuroplastizität gilt daher als gemeinsamer Nenner der sich langsam ausbildenden antidepressiven Wirkung der Antidepressiva, wobei, wie in Abbildung 7 dargestellt, die initialen Angriffspunkte sehr unterschiedlich sein können. Ebenfalls unterschiedlich können die einzelnen Signalwege sein, die dann letztlich, bevor es zu den eigentlichen histologisch fassbaren Veränderungen kommt, den Transkriptionsfaktor CREB (cAMP response element-binding protein) als gemeinsame Endstrecke aktivieren. Der aktivierte Transkriptionsfaktor führt dann seinerseits zur Synthese neurotropher Faktoren und damit zum Anstoßen von Neuroplastizitätsphänomenen. Die gemeinsame Endstrecke erklärt, dass sich die im primären Wirkungsmechanismus sehr unterschiedlichen Antidepressiva in der finalen antidepressiven Wirksamkeit eher nicht unterscheiden. In dieses Schema passen sehr gut auch Tianeptin, das zusätzlich zu Effekten in der serotoninergen Neurotransmission deutliche Effekte auf Neuroplastizitätsmechanismen über eine direkte Modulation glutamaterger Mechanismen aufweist, und der glutamaterge (NMDA) Antagonist Ketamin mit ebenfalls typischen Neuroplastizitätseffekten.

Abb. 6. Ebenen der synaptischen Plastizität. Synaptische Plastizität ist ein wichtiger Komplex von Mechanismen, mit denen die individuelle Empfindlichkeit von Synapsen programmiert wird und mit denen synaptische Funktion adaptiv auf Perioden von Über- bzw. Unteraktivität reagieren kann.
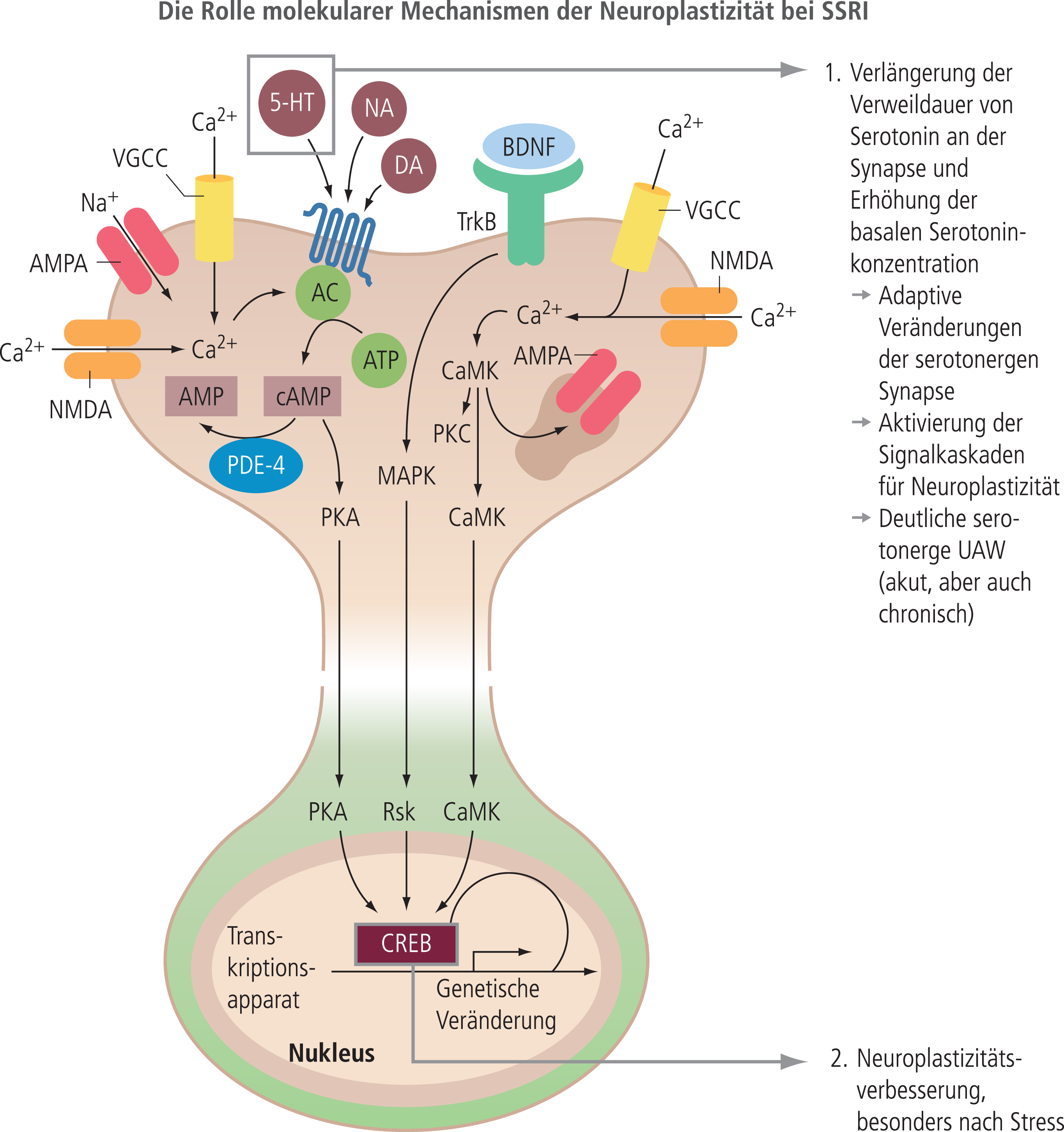
Abb. 7. Aktivierung von Neuroplastizität als gemeinsame Endstrecke der Antidepressiva-Wirkung. Nach Aktivierung von unterschiedlichen Targets an der Zelloberfläche führen verschiedene intrazelluläre Signalwege letztlich zur Aktivierung von CREB, einem Transkriptionsfaktor, der zur Aktivierung vieler neurotropher Faktoren führt. Bei den SSRI wird die Signalkaskade durch die Erhöhung der synaptischen Serotoninkonzentration angestoßen, die aber auch gleichzeitig Ursache der vielen serotoninergen UAW der SSRI ist (nach Pittenger et al., Neuropsychopharmacology 2008). Während SSRI glutamaterge Mechanismen nur indirekt (über die Aktivierung glutamaterger Neurone) beeinflussen können, hat Tianeptin einen direkten Effekt auf die glutamatergen Signalkaskaden, unter anderem über einen Effekt auf die Verfügbarkeit von AMPA- und NMDA-Rezeptoren (Müller, Psychopharmakotherapie 2016;23:102–17).
Während damit der akute Wirkungsmechanismus eher nicht von Bedeutung für die finale antidepressive Wirksamkeit ist, scheint er für den individuellen Patienten doch eine wichtige Rolle zu spielen dahingehend, dass der Weg bis zur Verbesserung der synaptischen Plastizität individuell unterschiedlich sein kann. Daher ist es klinischer Standard, bei Nichtansprechen auf ein bestimmtes Antidepressivum alternativ eine Substanz mit unterschiedlichem biochemischem Angriffspunkt zu wählen. Die akuten Angriffspunkte der Antidepressiva (Wiederaufnahmetransporter, Rezeptoren, Enzyme) spielen aber eine sehr große Rolle für die große und sehr unterschiedliche Anzahl von unerwünschten Arzneimittelwirkungen (UAW) der individuellen Antidepressiva, die sehr häufig den Patienten sehr belasten und therapielimitierend sein können. Die Wahl eines bestimmten Antidepressivums für einen bestimmten Patienten ist daher auch sehr deutlich von der zu erwartenden Verträglichkeit im Hinblick auf die UAW, bezogen auf den akuten Wirkungsmechanismus bestimmt.
Auf die praktische Anwendung von Antidepressiva gehen wir in der nächsten Folge ein.
Interessenkonflikterklärung
WEM: Vortrags- und Beraterhonorare von Neuraxpharm und Schwabe
GL: Keine Interessenkonflikte bezogen auf diesen Beitrag
Weiterführende Literatur
1. Bauer M. Neurobiologie und Therapie depressiver Erkrankungen. 5. Auflage. Bremen: Uni-Med, 2016.
2. Benkert O, Hippius H, et al. Psychiatrische Pharmakotherapie. 11. Auflage. Heidelberg: Springer, 2017.
3. Müller WE, Eckert A. Psychopharmakotherapie – pharmakologische Grundlagen. In: Möller HJ, Laux G, Kapfhammer HP (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. 5. Auflage, Band 2, Kapitel 31. Berlin: Springer, 2017.
Prof. Dr. Walter E. Müller, Höhenstraße 49A, 67550 Worms
Prof. Dr. med. Dipl.-Psych. Gerd Laux, Institut für Psychologische Medizin (IPM), Nussbaumstraße 9, 83564 Soyen, E-Mail: ipm@ipm-laux.de
Curriculum psychopharmacology/pharmacotherapy for psychiatric residents. Part 1: General psychopharmacotherapy, general pharmacology, and pharmacology of antidepressants
Psychopharmakotherapie 2017; 24(06):276-282