Walter E. Müller, Worms
Aripiprazol gehört zur Gruppe der atypischen Antipsychotika (atypischen Neuroleptika) und wurde Anfang der Achtzigerjahre zur Therapie der Schizophrenie eingeführt. In Deutschland ist es heute als Abilify® zur Akut- und Erhaltungstherapie der Schizophrenie und zur Akut- und Erhaltungstherapie manischer Phasen im Rahmen einer Bipolar-I-Erkrankung zugelassen [15]. Im Vergleich zu den meisten anderen atypischen Antipsychotika zeichnet sich Aripiprazol aufgrund seines besonderen Wirkungsmechanismus mit einem günstigen Spektrum von unerwünschten Arzneimittelwirkungen (UAW) aus, mit fast keiner Gewichtszunahme oder sonstigen metabolischen UAW, keinem Prolactinanstieg, geringen extrapyramidalmotorischen UAW (EPS) und keinen EKG-Veränderungen. Dies wurde auch besonders bei den klinischen Untersuchungen zur oralen Erhaltungstherapie als außergewöhnlicher Vorteil bewertet. Aripiprazol war hier im Hinblick auf den Endpunkt „Absetzen der Therapie wegen fehlender Wirkung bzw. wegen UAW der Plazebo-Gruppe“ hochsignifikant überlegen, schnitt hier auch besser ab als Haloperidol und war vergleichbar mit oralem Olanzapin [12, 23, 39, 41].
Trotz günstigem UAW-Profil hat auch die orale Erhaltungstherapie mit Aripiprazol ein nicht zu unterschätzendes Compliance-Problem, ähnlich wie bei anderen atypischen Antipsychotika, da Compliance nicht nur durch Verträglichkeit sondern auch durch viele andere Faktoren beeinflusst wird. Eine Möglichkeit der Compliance-Verbesserung ist der Einsatz von injizierbaren Depot-Arzneiformen für typische, besonders aber auch atypische Antipsychotika, die Dosierungsintervalle zwischen zwei und vier Wochen erlauben [3, 10, 11, 17, 40, 45, 63]. Aripiprazol bietet sich aufgrund seiner guten Wirksamkeit und Verträglichkeit von Anfang an besonders für eine solche Arzneiform an, wie sie dann Mitte 2014 unter dem Namen Abilify Maintena® in Deutschland in die Therapie eingeführt werden wird [16, 36].
Die Zulassung beruht auf zwei großen Langzeitstudien [22, 43]. Die besonderen pharmakologischen und pharmakokinetischen Eigenschaften, die dieser Anwendung zugrunde liegen, werden im Folgenden ausführlich dargestellt.
Die galenische Formulierung
Ziel einer Depotarzneiform ist die Aufrechterhaltung eines therapeutischen Plasmaspiegels über eine längere Zeit (in der Regel Wochen bis wenige Monate) durch eine einzelne Arzneimittelgabe. Die damit verbundenen langen Dosierungsintervalle sollen die regelmäßige Einnahme erleichtern und die Compliance verbessern, was besonders für die Depot-Antipsychotika gilt.
Depotarzneien können nicht mit jedem Wirkstoff hergestellt werden. Wesentliche Voraussetzung ist eine relativ geringe Tagesdosis (in der Regel im unteren Milligrammbereich), die meist mit einer hohen Affinität zur Zielstruktur einhergeht. Der Grund ist darin zu sehen, dass die Menge an Wirkstoff, die in eine Depotarzneiform gepackt werden kann, begrenzt ist. Da die Eliminationsprozesse für einen Wirkstoff, wenn er erst einmal im Blut ist, bei oraler Einnahme und bei Gabe als Depot (z.B. als Injektion) nicht unterschiedlich sind, kann die benötigte Menge in einer ersten vereinfachten Annäherung aus der oralen Erhaltungsdosis abgeschätzt werden, wenn der Wirkstoff nicht einen sehr großen First-Pass-Metabolismus hat. Beim Aripiprazol passt dies relativ gut: 400 mg Depot über vier Wochen im Vergleich zu einer täglichen Erhaltungsdosis von 15 mg. Hingegen würde eine Depotform für atypische Antipsychotika mit Tagesdosen von mehreren Hundert Milligramm (z.B. Clozapin, Sulpirid, Amisulprid) dermaßen große Dosen beim Depot erfordern, dass eine Verabreichung ins Gewebe nicht mehr möglich wäre.
Die galenische Herausforderung an einen Wirkstoff für die Depotzubereitung ist die Notwendigkeit, eine Zubereitung zu finden, die eine gleichmäßige Abgabe des Wirkstoffs aus der Arzneiform ins Gewebe beziehungsweise die Blutbahn über die gewünschte Zeit im Sinne einer Kinetik nullter Ordnung ermöglicht, also die Freisetzung der gleichen Menge des Wirkstoffs an jedem Tag als Voraussetzung für gleichmäßige Plasmaspiegel innerhalb des Dosierungsintervalls. Es wäre fatal, wenn die freigesetzte Menge von der vorhandenen Menge Wirkstoff bestimmt würde (Kinetik 1. Ordnung), dann hätte man am Anfang viel zu hohe und am Ende viel zu niedrige Plasmaspiegel.
Diese Anforderungen für einen Wirkstoff als Depotarzneiform erklären, dass für die atypischen Antipsychotika nur eine begrenze Zahl an Depot-Präparaten zur Verfügung steht (Tab. 1). Welche galenischen Tricks letztlich gewählt werden, ist von den physikochemischen Eigenschaften des Wirkstoffs abhängig. Eine wichtige Voraussetzung für die langsame Bereitstellung ist eine kontrollierte Löslichkeit, die über verschiedene Strategien erreicht werden kann (Veresterung, Salzbildung, Herstellung sich langsam lösender Mikropartikel) (Tab. 1). Im Fall von Aripiprazol-Depot wurden durch Lyophilisation gewonnene sich langsam lösende Mikropartikel verwendet [44]. Für die Anwendung am Patienten ist der Weg eher von untergeordneter Bedeutung, was zählt ist das Resultat: der gleichmäßige Plasmaspiegel [32].
Tab. 1. Atypische Depot-Antipsychotika am Markt und Vergleich zu Haloperidol-Depot
|
Substanz |
Haloperidol |
Risperidon |
Olanzapin |
Paliperidon |
Aripiprazol |
|
Handelsname |
Haldol-Janssen Decanoat® |
Risperdal-Consta® |
Zypadhera® |
Xeplion® |
Abilify Maintena® |
|
Wirkstoff |
Haloperidol-Decanoat |
Risperidon |
Olanzapin-Pamoat |
Paliperidon-Palmitat |
Aripiprazol |
|
Freisetzungs-mechanismus |
Prodrug: Hydrolyse durch Esterasen |
Mikropartikel: Freisetzung durch langsames Auflösen des Wirkstoffs |
Salz: langsame Dissoziation zu Olanzapin und Pamoasäure |
Prodrug: Hydrolyse durch Esterasen |
Mikropartikel: Freisetzung durch langsames Auflösen des Wirkstoffs |
|
Formulierung |
Lösung Sesamöl |
Wässrige Suspension |
Wässrige Suspension |
Wässrige Suspension mit Nanokristallen |
Trockenpulver für wässrige Lösungen |
|
Dosierungsintervall |
4 Wochen |
2 Wochen |
2–4 Wochen |
4 Wochen |
4 Wochen |
Der Weg zur langsamen Freisetzung ist unterschiedlich. Haloperidol und Paliperidol erreichen dies durch Veresterung mit einer Fettsäure, Olanzapin durch Salzbildung mit Pamoasäure, Risperidon und Aripiprazol durch Verkapselung in Mikropartikel [16, 41].
Pharmakokinetik und Biotransformation
Die Pharmakokinetik einer Depotarzneiform wie bei Aripiprazol-Depot setzt sich aus zwei sehr unterschiedlichen, aber letztlich zusammenspielenden Mechanismen zusammen. Die Abgabe ins Plasma wird von dem in der Regel intramuskulär injizierten Depot und seiner langsamen Lösung bestimmt, die Abnahme des Wirkstoffs aus dem Blut hängt von den klassischen Eliminationsmechanismen ab, im Falle des Wirkstoffs Aripiprazol vom hepatischen Metabolismus, und folgt den gleichen Kriterien wie bei einer oralen Einnahme. Im Fließgleichgewicht nach mehrfacher Anwendung des Depots (Abb. 1) spielen beide Mechanismen zusammen. Die über den Metabolismus täglich ausgeschiedene Menge wird durch die neu aus dem Depot gelöste Menge Wirkstoff ersetzt, sodass über viele Wochen ein relativ stabiler Plasmaspiegel erreicht wird (Abb. 1).
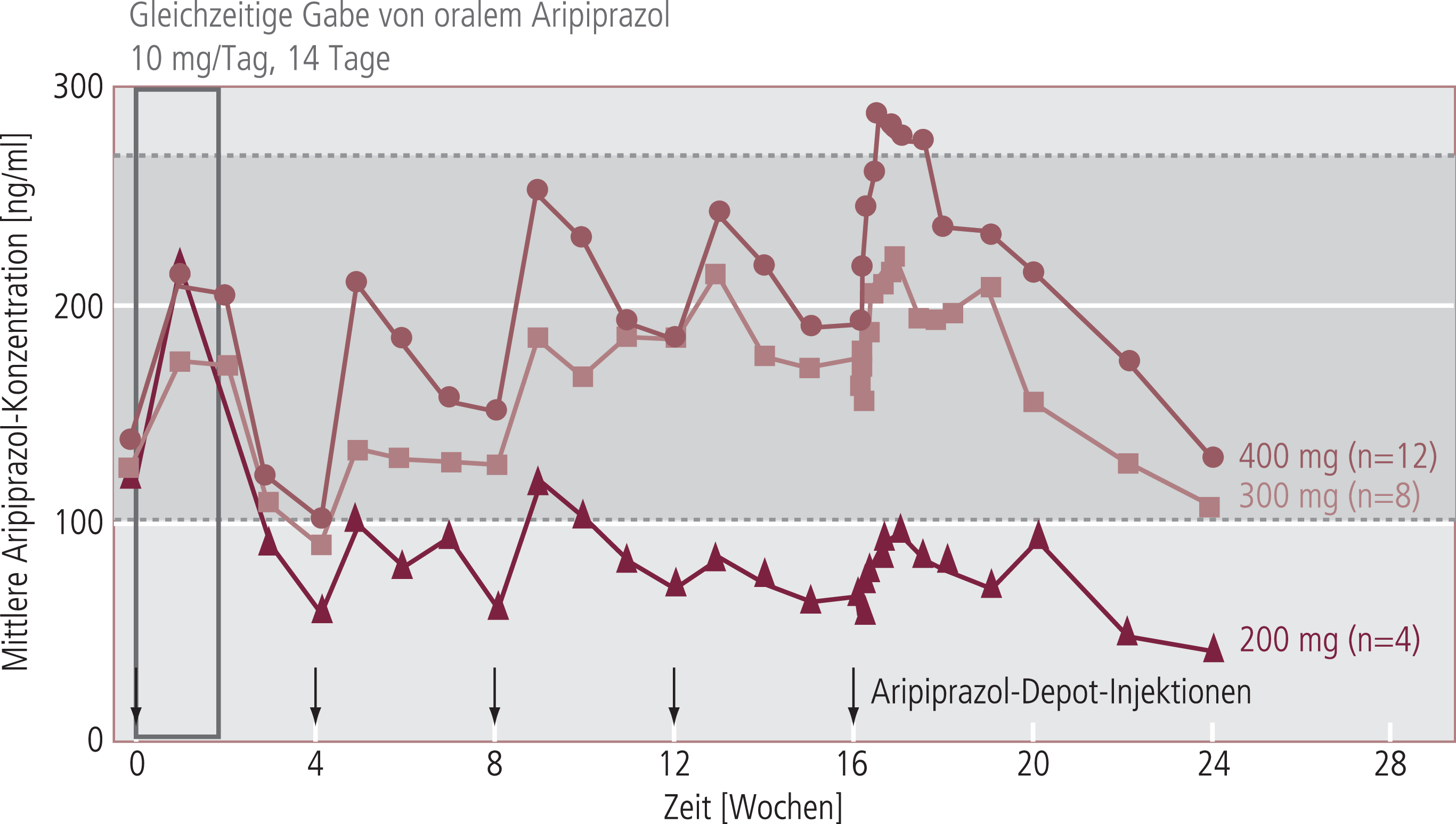
Abb. 1. Pharmakokinetik von Aripiprazol-Depot an schizophrenen Patienten [29]. Aripiprazol wurde in drei unterschiedlichen Dosen insgesamt 5-mal über 16 Wochen gegeben. Um den angestrebten Plasmaspiegelbereich (schraffierte Fläche) schneller zu erreichen, erhielten die Patienten in den ersten 14 Tagen zusätzlich 10 mg/Tag Aripiprazol oral. Die Darstellung ist modifiziert nach [29].
Die Bedeutung der Pharmakokinetik des Depots kommt an zwei Punkten der Zeitachse zum Tragen. In den ersten Wochen nach der initialen Injektion ist die Aripiprazol-Freigabe aus dem Depot noch nicht ausreichend, um allein den gewünschten Plasmaspiegel von etwa 100 bis 200 ng/ml zu erreichen, sodass eine zusätzliche orale Einnahme von 10 mg Aripiprazol empfohlen wird (Abb. 1). Nach Absetzen der Depot-Injektionen wird der Plasmaspiegel zunächst nicht durch die klassische Eliminationshalbwertszeit beschrieben, sondern durch die Freigabe aus dem Depot (Tab. 2). Diese Halbwertszeit liegt bei etwa 47 Tagen (400-mg-Depot) beziehungsweise 40 Tagen (300-mg-Depot). Erst ganz am Ende, wenn kein Wirkstoff mehr aus dem Depot freigesetzt werden kann, folgt der Plasmaspiegelverlauf den gleichen pharmakokinetischen Kriterien wie nach oraler Einnahme.
Tab. 2. Pharmakokinetische Eckdaten für den Plasmaspiegelverlauf von Aripiprazol nach Gabe von Aripiprazol-Depot (400 mg bzw. 300 mg) [nach 29]
|
Pharmakokinetische Parameter |
Aripiprazol-Depot 400 mg |
Aripiprazol-Depot 300 mg |
|
Cmax [ng/ml] |
316 |
269 |
|
tmax [Tage] |
7,1 |
6,5 |
|
AUCτ [µg×h/ml] (SD) |
163 (88,8) |
140 (58,4) |
|
t½ [Tage] (SD) |
46,5 (10,8) |
29,9 (8,0) |
|
Cmin [ng/ml] (SD) |
212 (113) |
156 (67,7) |
Im Gegensatz zur sonstigen Bedeutung der Eliminationsprozesse für diese Konstanten werden sie bei Aripiprazol-Depot primär über die Abgabe aus dem Depot ins Plasma bestimmt. Basierend auf der AUCSS (Fläche unter der Plasmakonzentrations-Zeit-Kurve im Steady-State) liegt das orale Äquivalent von 400 mg Aripiprazol-Depot bei 21,2 mg und von 300 mg Aripiprazol-Depot bei 15,9 mg; SD: Standardabweichung
Die schon lange bekannten pharmakokinetische Daten zu oralem Aripiprazol sind in Tabelle 3 zusammengefasst. Die Substanz wird relativ langsam resorbiert mit einer Zeit zum maximalen Plasmaspiegel von etwa drei bis fünf Stunden und verteilt sich zeitlich ähnlich in die Gewebe, einschließlich des Gehirns, hat aber ein relativ kleines Verteilungsvolumen von 4,9 l/kg, was gegen eine hohe Anreicherung im Gewebe spricht. Aripiprazol wird hauptsächlich metabolisch eliminiert unter Beteiligung von Cytochrom P450(CYP)-2D6 und CYP3A4 (Abb. 2). Hauptmetabolit ist Dehydroaripiprazol, das analog Aripiprazol wirksam ist. Nur geringe Mengen werden unverändert über die Fäzes und den Urin ausgeschieden. Die terminale Elimination ist mit einer Halbwertszeit von etwa vier Tagen sehr langsam. Dadurch benötigt es auch rund 20 Tage, bis nach Beginn einer oralen Therapie ein Steady-State des Plasmaspiegels eingestellt ist, beziehungsweise werden 20 Tage benötigt, bis sich nach Dosisänderung (Reduktion oder Erhöhung) das neue Fließgleichgewicht wieder eingestellt hat [6, 9, 52]. Der aktive Metabolit Dehydroaripiprazol (Abb. 2) wird auch sehr langsam mit einer mittleren Halbwertszeit von etwa 130 Stunden eliminiert [9]. Seine Plasmakonzentration ist etwa ein Drittel der Konzentration von Aripiprazol (Abb. 3).
Tab. 3. Die wichtigsten pharmakokinetischen Daten zu oralem Aripiprazol [15, 52]
|
Empfohlene Dosis |
10–15 mg/Tag (maximal 30 mg) |
|
Terminale Halbwertszeit (t ½) |
100 h |
|
150 h |
|
75 h |
|
Zeit bis Fließgleichgewicht (Steady-State) |
20 Tage |
|
Maximale Plasmaspiegel nach oraler Einnahme |
3–5 h |
|
Bioverfügbarkeit |
87% |
|
Verteilungsvolumen |
4,9 l/kg |
|
Metabolismus |
Über CYP2D6 und CYP3A4 |
|
Hauptmetabolit (aktiv) |
Dehydroaripiprazol |
CYP: Cytochrom P450
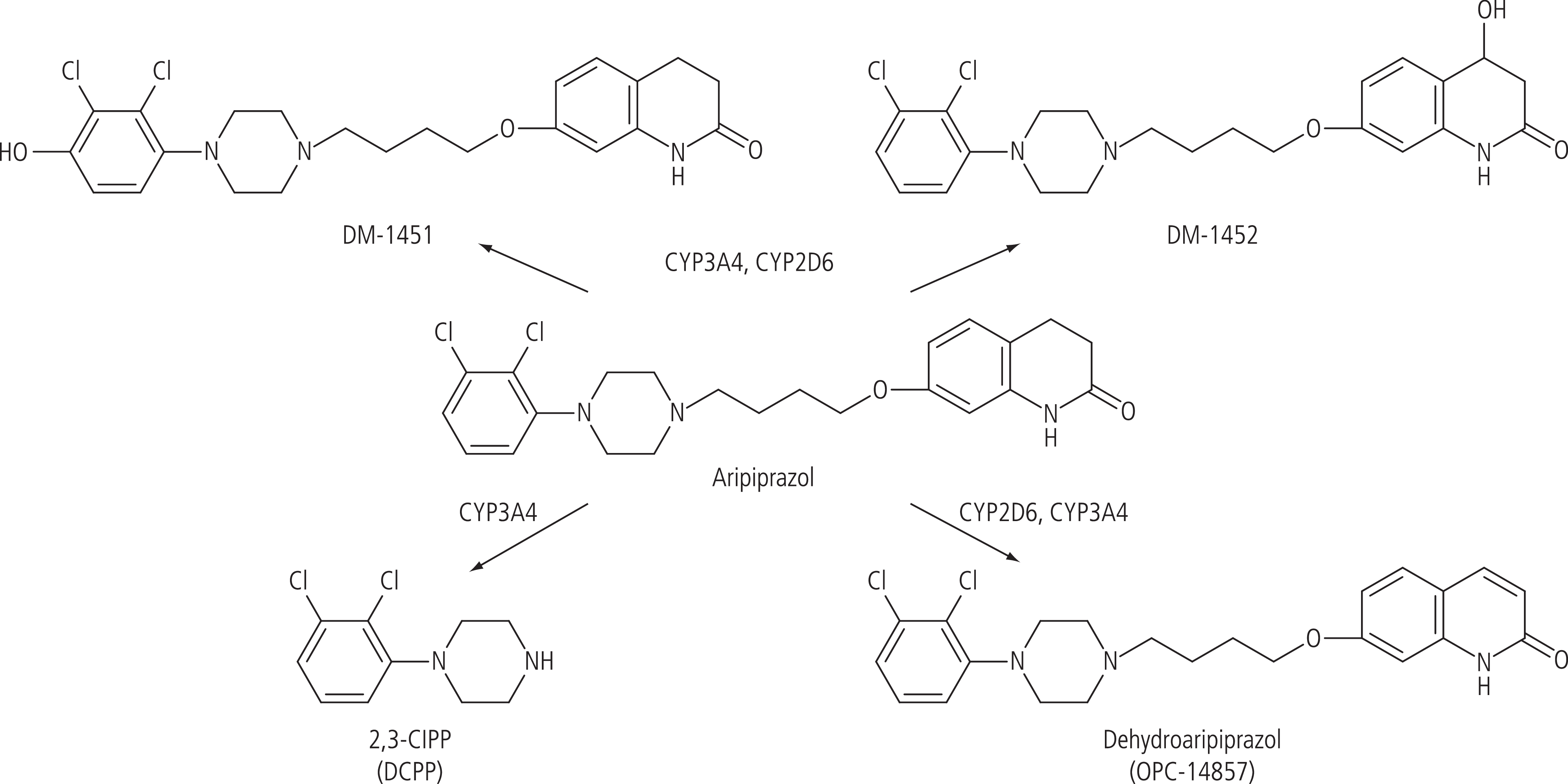
Abb. 2. Schema des Metabolismus von Aripiprazol am Menschen. Hauptmetabolit ist Dehydroaripiprazol, das als aktiver Metabolit zur Wirkung beiträgt. Seine Plasmaspiegel erreichen etwa ein Drittel der Plasmaspiegel der Muttersubstanz [mod. nach 9].
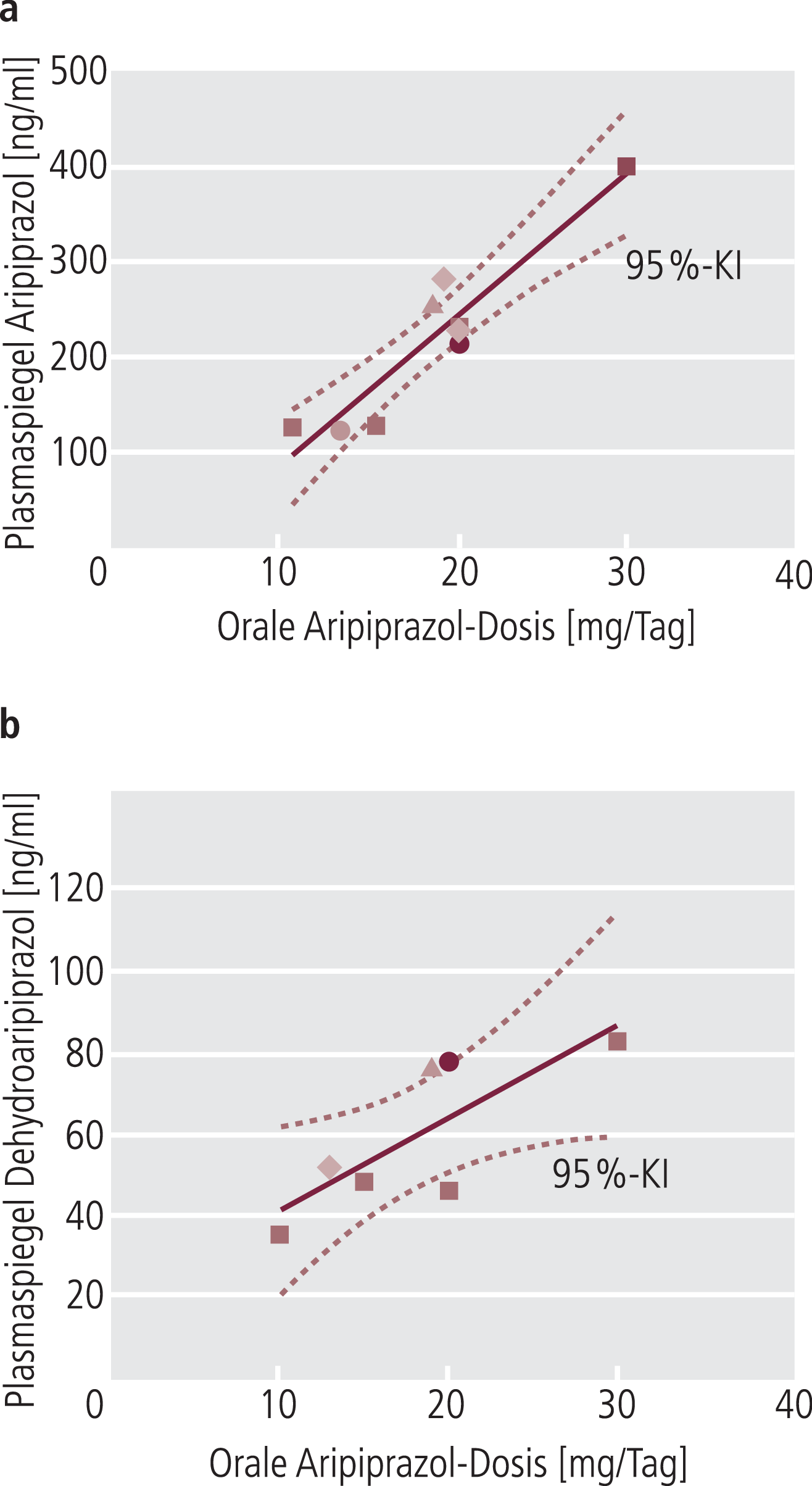
Abb. 3. Zusammenfassung [52] der Daten aus verschiedenen Untersuchungen zur Beziehung zwischen oraler Dosis von Aripiprazol und den gemessenen Plasmaspiegeln der Muttersubstanz (3a) sowie dem wirksamen Hauptmetaboliten Dehydroaripiprazol (3b). Jeder der Punkte stellt die Mittelwerte einer unabhängigen Untersuchung dar. Die gestrichelten Linien sind die 95%-Konfidenzintervalle (KI) und die gestrichenen Linien die berechneten linearen Korrelationen. Die Darstellungen sind modifiziert nach [52].
Aufgrund des relativ kleinen Verteilungsvolumens von Aripiprazol besteht im Mittel eine sehr gute Beziehung zwischen oraler Dosis und Plasmaspiegel, was auch für Dehydroaripiprazol gilt (Abb. 3). Trotzdem ist aufgrund der bekannten großen interindividuellen Varianz der beiden Enzyme CYP2D6 und CYP3A4 die interindividuelle Schwankung der Plasmaspiegel bei gegebener Dosis erheblich. Die dadurch entstehenden Probleme, Dosis beziehungsweise Plasmaspiegel mit der Wirkung zu korrelieren, können deutlich reduziert werden, wenn die Plasmaspiegel von Aripiprazol und Dehydroaripiprazol als Summe betrachtet werden [35].
In den ursprünglichen Zulassungsstudien hat man maximale therapeutische Wirkung von Aripiprazol bei schizophrenen Patienten mit einer Tagesdosis zwischen 10 mg und 20 mg gesehen [52], was Basis für die aktuellen Therapieempfehlungen ist [15]. Die bei diesen Dosen erreichten Plasmakonzentrationen (Abb. 3a) waren auch der angestrebte Dosisbereich bei der Entwicklung von Depot-Aripiprazol (Abb. 1). Dieser Bereich wurde später auch durch D2-Rezeptor-Okkupationsstudien belegt, wo man am Menschen maximale Effekte (über 80% Okkupation) ab einem Plasmaspiegelbereich von etwa 100 ng/ml gesehen hat (Abb. 4) [62], der bereits im Mittel von einer oralen Tagesdosis von rund 10 bis 15 mg erreicht wird (Abb. 3a).
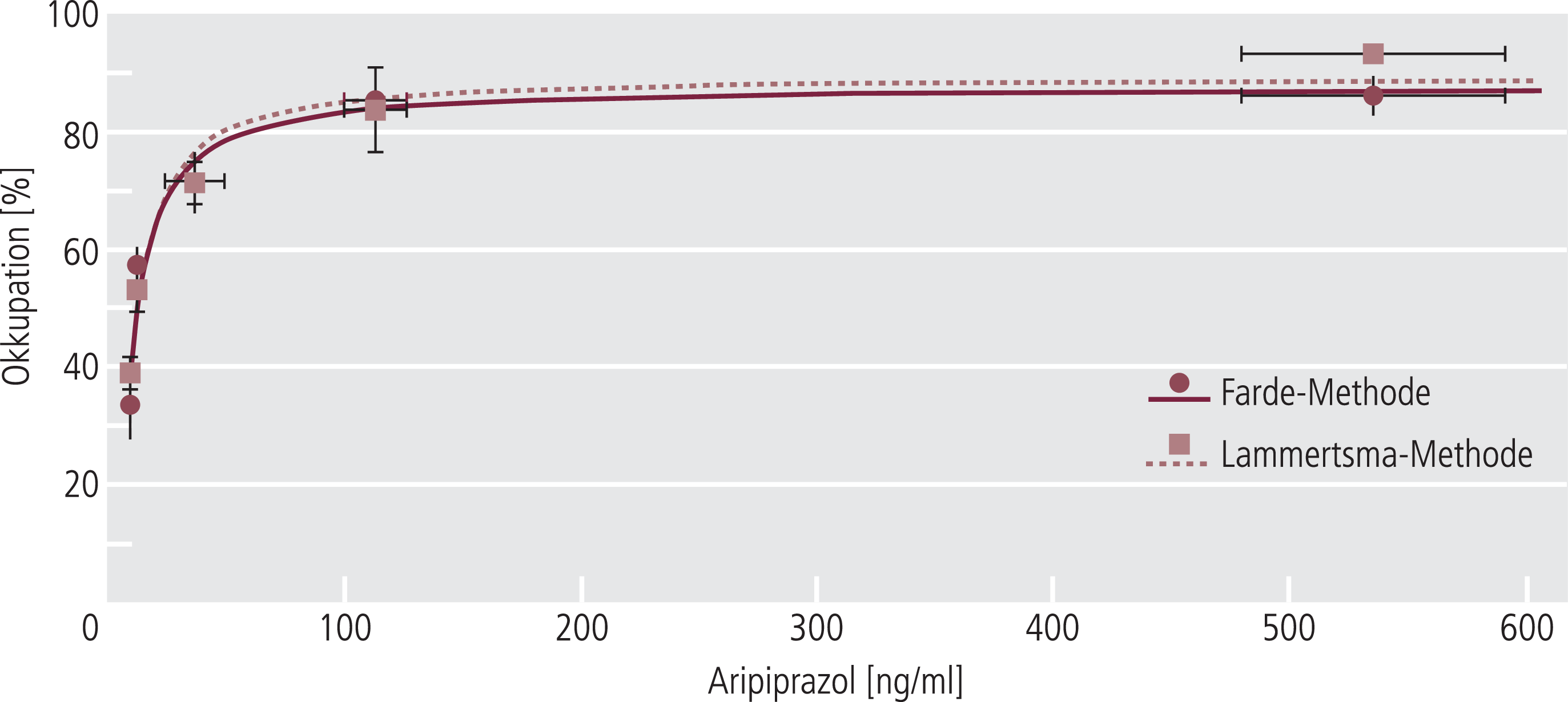
Abb. 4. Die Beziehung von Plasmakonzentration von Aripiprazol und der Okkupation des primären pharmakologischen Targets (Dopamin-D2-Rezeptor im Striatum). Maximale Effekte sind bereits bei Konzentrationen etwas über 100 ng/ml erreicht. Die Untersuchung wurde an gesunden Probanden mit der Positronenemissionstechnik unter Verwendung des Radioliganden [11C]-Racloprid durchgeführt [62]. Die Rezeptorokkupation wurde nach zwei unterschiedlichen Methoden berechnet [62].
Es stellt sich natürlich die Frage, welche dieser allgemeinen pharmakokinetischen Parameter von oralem Aripiprazol für das Depot relevant sind. Wie bereits erwähnt, wird bei Aripiprazol-Depot der Plasmaspiegel durch die Lösung aus dem intramuskulär injizierten Depot bestimmt. Die Menge, die das Depot pro Tag abgeben muss, wird dagegen durch die Menge von Aripiprazol im Plasma bestimmt, die täglich eliminiert wird (Abb. 1). Dies ist natürlich eine Folge des oben beschriebenen hepatischen Metabolismus, sodass auch bei der Anwendung des Depots der primäre wirksame Metabolit Dehydroaripiprazol Plasmaspiegel von etwa einem Drittel der Plasmaspiegel der Muttersubstanz erreicht (Abb. 5). Dadurch dass der Plasmaspiegel beim Depot durch die Menge erreicht wird, die pro Tag aus dem Depot freigesetzt wird, und die Menge ersetzen soll, die durch den normalen Metabolismus eliminiert wird, ist der Plasmaspiegel bei Anwendung von Aripiprazol-Depot genauso empfindlich für pathopysiologische und. pharmakologische Veränderungen des Eliminationsprozesses. Das System ist sogar empfindlicher, da das banale Mittel einer Dosisanpassung der Muttersubstanz bei der oben erwähnten mittleren Halbwertszeit (über 40 Tage) wenig akuten Erfolg verspricht. Daher sollten Patienten mit bekanntem schnellem oder langsamem Metabolismus (Tab. 1) gegebenenfalls mit der nächst höheren beziehungsweise nächst niedrigeren Dosis behandelt werden. Langfristige Komedikation mit CYP2D6- oder CYP3A4-Inhibitoren beziehungsweise -Induktoren sollte vermieden werden. Kurzfristige Gabe (unter 14 Tage) ist möglicherweise aufgrund der Trägheit der Plasmaspiegelverläufe (Abb. 1 und 5) unter Kontrolle möglich [16]. Ähnlich wie bei oralem Aripiprazol ist der Plasmaspiegel sonst relativ unempfindlich, sodass bei älteren Patienten, Rauchern, Männer oder Frauen, Patienten mit unterschiedlicher ethnischer Angehörigkeit und selbst bei Patienten mit Störungen der Nieren- oder Leberfunktion keine generellen Dosisanpassungen empfohlen werden [16, 59].
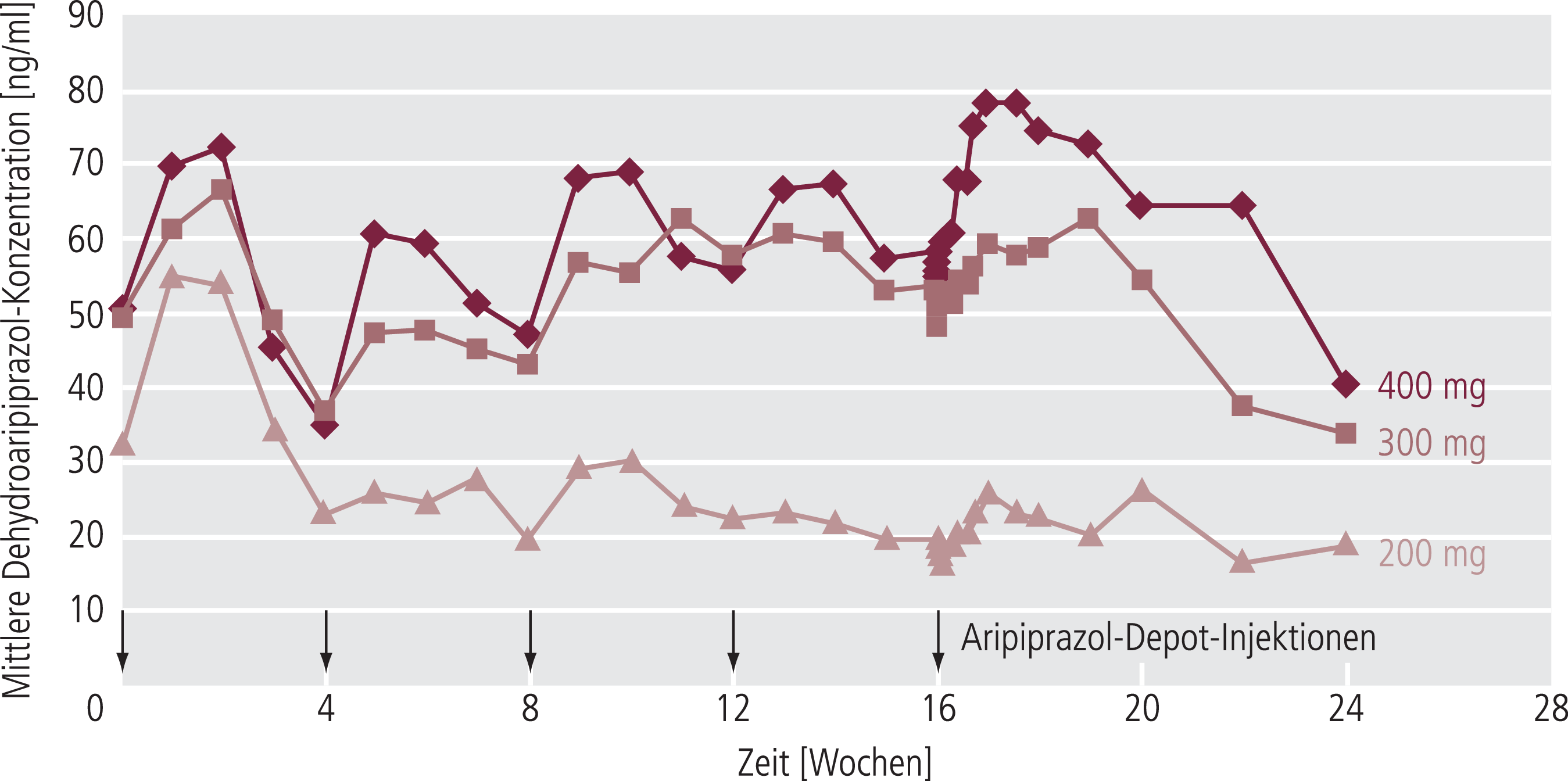
Abb. 5. Fast deckungsgleicher Verlauf der Plasmaspiegel von Dehydroaripiprazol zu den Plasmaspiegeln der Muttersubstanz (Abb. 1). Auch hier betragen die Werte etwa ein Drittel der Werte für Aripiprazol. Die Darstellung ist modifiziert nach [29].
Pharmakologie
Die ersten Depot-Antipsychotika waren typische ältere Neuroleptika (Fluphenazin, Flupentixol, Haloperidol) – üblicherweise wie bei Haloperidol-Decanoat (Tab. 1) durch Veresterung mit längerkettigen Fettsäuren in eine sich nur langsam lösende Form überführt. Trotz guter Wirksamkeit waren sie mit deutlichen Akzeptanzproblemen verbunden, besonders auch wegen der nicht unerheblichen UAW der älteren Antipsychotika. Es lag daher nahe, auch für einige atypische Antipsychotika Depot-Arzneiformen zu entwickeln mit dem Ziel, deren bessere Verträglichkeit auch bei der Erhaltungstherapie und Rezidivprophylaxe auszunutzen (Tab. 1). Aripiprazol-Depot ist das jüngste dieser Depot-Atypika und bringt auch durch sein innerhalb der Gruppe der Atypika günstiges UAW-Profil gute Voraussetzungen für diese Anwendung mit. Grundlage für das besonders günstige UAW-Profil von Aripiprazol sind seine besonderen pharmakologischen Eigenschaften und als Grundlage dafür sein besonderes Rezeptorbindungsprofil.
Antipsychotischer Wirkungsmechanismus: Partieller Agonismus bzw. partieller Antagonismus an Dopamin-D2-, D3-Rezeptoren
Die besondere Pharmakologie von Aripiprazol ist schon seit Jahren bekannt und soll daher hier nur relativ kurz zusammengefasst werden. Der interessierte Leser sei auf verschiedene Übersichtsarbeiten zur Pharmakologie dieses interessanten Atypikums verwiesen [12, 26, 38, 51]. Alle typischen und atypischen Antipsychotika wirken über eine Blockade von zentralen Dopamin-Rezeptoren vom D2- und D3-Typ antipsychotisch, wobei besonders, aber nicht ausschließlich Rezeptoren in den mesolimbischen Projektionsgebieten eine große Rolle spielen. Die gleichzeitige Blockade dieser Rezeptoren in den Basalganglien und der Hypophyse erklärt die typischen UAW wie EPS und Prolactinanstieg, die man früher als nicht abtrennbar von der neuroleptischen Wirkung angesehen hat. Dieses Dogma wurde durch die Atypika – ausgehend vom Clozapin – durchbrochen, die alle weniger EPS und Prolactinanstieg (Ausnahme Amisulprid, mit Einschränkung Risperidon) zeigen als die alten Typika. Die Erklärung für die deutliche Reduktion der EPS ist darin zu sehen, dass die Atypika bei antipsychotischer Dosierung etwas geringer an D2-Rezeptoren im Striatum binden als in den für die antipsychotische Wirkung wichtigen mesolimbischen Gebieten [37]. Wird die Dosis des Atypikums gesteigert, dann treten wegen höherer Blockade der D2-Rezeptoren im Striatum (etwa über 80% Okkupation) auch bei den meisten Atypika vermehrt EPS auf. Die Mechanismen, über die die Reduktion von EPS bei den einzelnen Atypika erreicht wird, sind allerdings unterschiedlich. Entweder kann es aufgrund einer leichteren Verdrängbarkeit durch das im Striatum sowieso sehr hoch konzentrierte extrazelluläre Dopamin zu einer Bindungskompetition kommen (dies gilt für die sogenannten Fast-off-Substanzen [Clozapin, Quetiapin]), oder es kommt durch eine sehr starke gleichzeitige Blockade von 5-HT2A-Rezeptoren zu einer Desinhibition der nigrostriatalen dopaminergen Neuronen mit einer zusätzlichen erhöhten Dopamin-Freisetzung im Striatum (gilt zumindest auch für fast alle Atypika mit Ausnahme von Amisulprid) [37].
Aripiprazol ist hier die ganz große Ausnahme, da es diese Mechanismen nur bedingt bedient, mit sehr geringem Risiko für EPS und Prolactinerhöhungen, aber zweifelsfrei zu den Atypika zu zählen ist. Es hat die höchste Affinität zum D2-Rezeptor aller Atypika und wäre daher nach der alten Klassifikation ein hochpotentes Neuroleptikum (Tab. 4), und es okkupiert unter therapeutischen Bedingungen auch deutlich mehr als 80% der D2-Rezeptoren im Striatum (siehe Abb. 4). Dass Aripiprazol sich trotzdem atypisch verhält, liegt daran, dass die Substanz am D2- und D3-Rezeptor partialagonistische bzw. partialantagonistische Eigenschaften aufweist. Das bedeutet, dass es bei Bindung an den Rezeptor zwar agonistische Effekte auslöst, aber sehr viel schwächer, als dies ein voller Agonist kann. Der so genannte intrinsische bzw. agonistische Effekt von Aripiprazol beträgt etwa 25% des vollen Agonisten Dopamin, sodass es eher ein partieller Antagonist ist [8, 55, 57, 60, 61]. Der aktive Metabolit Dehydroaripiprazol verhält sich an humanen D2-Rezeptoren analog, während Dehydroaripiprazol an der Ratte ein reiner D2-Antagonist zu sein scheint [60].
Tab. 4. Halbmaximale Bindungskonstanten (als Ki in nmol/l) an relevante zentrale Rezeptoren für die in Tabelle 1 zusammengefassten Depot-Antipsychotika [12, 49]
|
Rezeptor |
Antipsychotikum |
||||
|
Haloperidol |
Risperidon |
Paliperidon |
Olanzapin |
Aripiprazol |
|
|
D1 |
210 |
620 |
670 |
31 |
265 |
|
D2 |
0,7 |
6 |
5 |
11 |
0,2 |
|
D3 |
2 |
14 |
7 |
49 |
0,8 |
|
D4 |
3 |
16 |
30 |
27 |
44 |
|
5-HT1A |
2600 |
420 |
587 |
>7000 |
4 |
|
5-HT2A |
45 |
0,2 |
0,2 |
4 |
3 |
|
5-HT2C |
1500 |
63 |
71 |
23 |
15 |
|
α1 |
6 |
2 |
14 |
19 |
57 |
|
α2 |
360 |
8 |
17 |
230 |
– |
|
H1 |
440 |
3 |
10 |
7 |
61 |
|
M1 |
>2000 |
>5000 |
>5000 |
2 |
>10000 |
Nimmt man an, dass man bei einem Affinitätsunterschied von einer Zehnerpotenz über der Targetaffinität (D2, D3) noch relevante Rezeptorokkupationen erwarten kann [37], sind nach diesen In-vitro-Daten für Aripiprazol noch der 5-HT1A- und der 5-HT2A-Rezeptor in vivo relevant
Unter bestimmten Bedingungen können nun solche partiellen Agonisten/Antagonisten funktionell entweder eher agonistische oder eher antagonistische Effekte auslösen. Zum einen hängt ihre Wirkung von der Konzentration des physiologischen Liganden (in diesem Fall Dopamin) ab. Bei hoher Ligandenkonzentration überwiegt die antagonistische Komponente, bei niedriger Ligandenkonzentration kommt bei Bindung an den Rezeptor die eigene, wenn auch schwache, agonistische Komponente zum Tragen. So hat man vermutet, dass bei der pathologisch erhöhten präsynaptischen Dopamin-Freisetzung, wie man sie als funktionell relevant in mesolimbischen Strukturen annimmt [20, 42], der antagonistische, antipsychotische Effekt überwiegt, während dort, wo Dopamin weniger vermehrt freigesetzt wird (z.B. Striatum), die eigenen agonistischen Effekte zum Tragen kommen und weniger EPS entstehen. Eine andere Erklärung sind die unterschiedlichen Effekte auf die synaptische Neurotransmission eines partiellen Agonisten bei unterschiedlicher Größe der Rezeptorreserve (Abb. 6). Auch damit können regional unterschiedliche Effekte von Aripiprazol auf die dopaminerge Neurotransmission möglich werden [38, 56]. Partieller D2-Agonismus ist damit zweifelsfrei der Hauptmechanismus für die geringe Häufigkeit von EPS bei der Anwendung von Aripiprazol in der Behandlung der Schizophrenie bei gleichzeitig guter antipsychotischer Wirkung und auch für den praktisch fehlenden Effekt auf die Prolactinfreisetzung [1]. Erklären lässt sich durch den partiellen Agonismus aber auch, dass in manchen Fällen die etwas schwächere D2-Rezeptor-Blockade allein für eine volle Reduktion der psychotischen Symptome nicht ausreicht, was besonders bei Patienten, die mit hohen Dosen anderer Antipsychotika behandelt werden, Probleme bereiten kann [58].
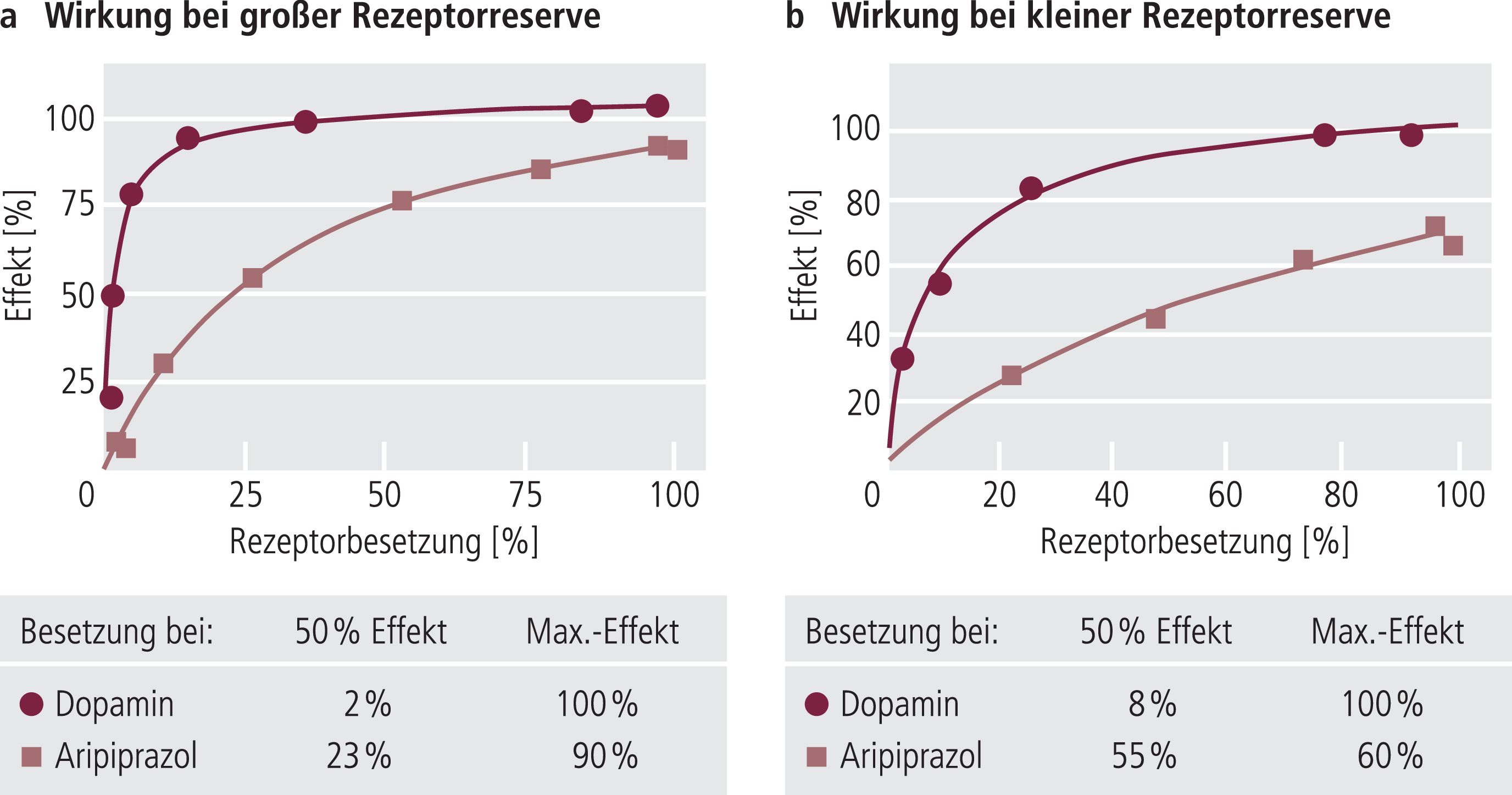
Abb. 6. Wirkung bei unterschiedlicher Rezeptorreserve. Bei großer Rezeptorreserve benötigt der volle Agonist Dopamin nur etwa 20% der Rezeptoren, um im vorgegebenen System volle Wirkung auszulösen. Der partielle Agonist benötigt für den gleichen Effekt 100% der Rezeptoren, da er ja an jedem Rezeptor allein weniger Effekt hat. Bei 100% Okkupation bekommt man aber auch mit dem partiellen Agonisten Aripiprazol die volle Wirkung. Benötigt allerdings schon der volle Agonist Dopamin alle vorhandenen Rezeptoren für volle Wirkung (kleine Rezeptorreserve), kann Aripiprazol selbst bei 100% Okkupation das System nicht voll aktivieren, mit dem Resultat eines partiellen Antagonismus [38].
Effekte an Serotonin-Rezeptoren
Aripiprazol bindet neben den Dopamin-Rezeptoren auch an eine Reihe anderer Neurorezeptoren (Tab. 4). Allerdings ist die Affinität zu allen anderen Systemen deutlich geringer. Nimmt man einen Unterschied der Bindungskonstante unter einer Zehnerpotenz als Grenzwert für eine pharmakologische Relevanz unter der Therapie an [37], dann ist bei Dosierungen von Aripiprazol im therapeutische Bereich (bis 30 mg pro Tag) gerade noch an den 5-HT1A-, den 5-HT2A- und mit Einschränkung an den 5-HT2C-Rezeptoren mit einer relevanten Bindung von Aripiprazol zu rechnen. Diese theoretischen Überlegungen werden durch die Studie von Mamo et al. [30] bestätigt, wo bei schizophrenen Patienten mit Aripiprazol-Dosen bis 30 mg pro Tag D2-Rezeptor-Okkupationen von fast 90% erreicht wurden, gleichzeitig aber der 5-HT2A-Rezeptor nur zu etwa 50% und der 5-HT1A-Rezeptor nur zu knapp 20% okkupiert war. Damit sind diese Rezeptoren sicher nur begrenzt am Wirkungsprofil von Aripiprazol am Patienten beteiligt. Auf der anderen Seite ist pharmakologisch (in vitro und im Tierversuch) eine Beteiligung beider serotonerger Rezeptoren an den Effekten von Aripiprazol gut belegt [5, 53]. Darüber hinaus scheint die relativ geringe In-vivo-Bindung von Aripiprazol an den 5-HT1A-Rezeptor am Menschen [30] die reale Situation aufgrund methodischer Artefakte (relativ schwache Verdrängung des verwendeten Radioliganden vom 5-HT1A-Rezeptor durch partielle Agonisten wie Aripiprazol) zu unterschätzen. Damit ist es durchaus möglich, dass die gleichzeitige Blockade von 5-HT2A- und 5-HT1A-Rezeptoren bei Aripiprazol zu den atypischen Eigenschaften beiträgt – in Analogie zu vielen anderen atypischen Antipsychotika [37] (Abb. 7). Am 5-HT1A-Rezeptor verhält sich Aripiprazol wie das Anxiolytikum Buspiron, aber auch wie einige andere Atypika als partieller Agonist [21]. Über die mögliche Bedeutung dieses Effekts für zusätzliche Effekte von Aripiprazol auf Affekt und Kognition wird im nächsten Abschnitt näher eingegangen.
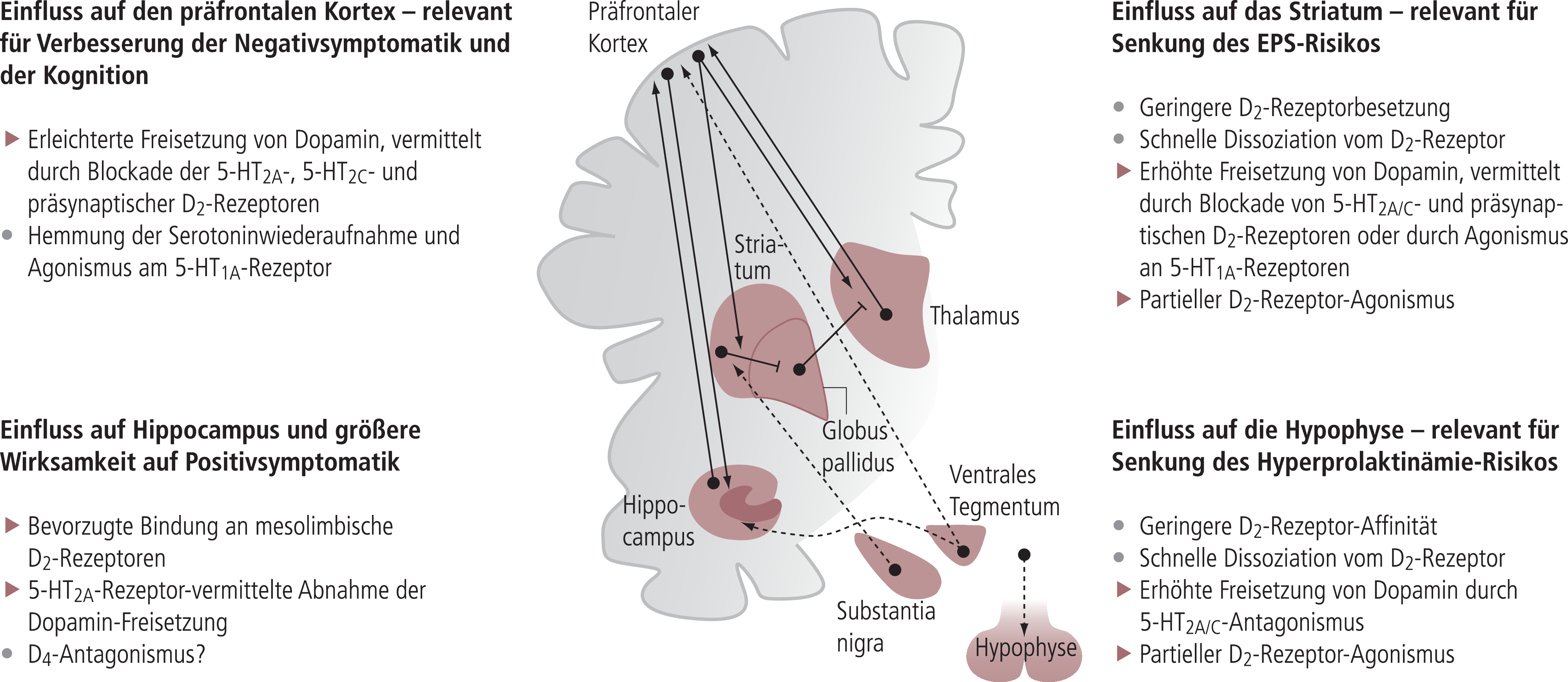
Abb. 7. Schematische Darstellung der Beeinflussung der relevanten zentralen dopaminergen Systeme durch Antipsychotika. In allen vier Arealen zeigen alle Antipsychotika als gemeinsamen Nenner eine Blockade von D2-Rezeptoren. Die Mechanismen, die die atypischen Antipsychotika von den typischen unterscheiden, sind dargestellt. Die Mechanismen, die auch für Aripiprazol gelten, sind mit einem roten Pfeil markiert [mod. nach 27]. EPS: Extrapyramidales Syndrom
Vielleicht sogar wichtiger als die Rezeptoren, an die Aripiprazol auch noch bindet, sind die allgemein für die UAW von Antipsychotika und Antidepressiva relevanten Neurorezeptoren, an die Aripiprazol unter therapeutischen Bedingungen nicht relevant (Tab. 4) bindet, was das sehr günstige UAW-Profil dieser Substanz innerhalb der Gruppe der atypischen Antipsychotika erklärt. Fehlende relevante Interaktion mit dem Histamin-H1-Rezeptor erklärt, dass die Substanz nicht sediert, und zusammen mit fehlender relevanter Bindung an den 5-HT2C-Rezeptor begründet diese Eigenschaft, warum Aripiprazol im Mittel zu keiner Gewichtszunahme führt und auch sonst im Hinblick auf metabolische Veränderungen (Glucose- und Lipidstoffwechsel) meist keine Unterschiede zu Plazebo gezeigt hat; Aripiprazol ist hier somit innerhalb der Gruppe der atypischen Antipsychotika gut positioniert, was auch in einer Metaanalyse herausgearbeitet wurde [46]. Fehlende M1- und Alpha1-Affinität und damit kaum anticholinerge und blutdrucksenkende beziehungsweise orthostatische Probleme zusammen mit der ausführlich erklärten geringen EPS- und Prolactin-Problematik runden dieses günstige UAW-Spektrum ab, was für die Langzeitanwendung in Form von Aripiprazol-Depot von großer Bedeutung ist [10, 11, 18, 39, 40, 45, 59].
Effekte auf Affekt und Kognition
Neben der Behandlung der Schizophrenie ist orales Aripiprazol auch für die Akutbehandlung manischer Phasen bei Bipolar-1-Patienten und zur Prävention manischer Phasen bei Bipolar-1-Patienten zugelassen [15] mit guter Wirkung und analog günstigem UAW-Profil [13, 33, 34]. Da eine antimanische Wirkung für praktisch alle untersuchten typischen und atypischen Antipsychotika gefunden wurde, kann man davon ausgehen, dass eine Blockade von D2- und D3-Rezeptoren auch bei der Aripiprazol-Anwendung bei Manie die pharmakologische Grundlage darstellt.
Eine Reihe von offenen, aber auch von Plazebo-kontrollierten Untersuchungen belegt eine gute klinische Wirksamkeit von Aripiprazol als Add-on-Medikation bei therapieresistenten Depressionen oder Angsterkrankungen [4, 24] allerdings ist Aripiprazol für keine der beiden Erkrankungen zugelassen. Auch in Experimenten an der Ratte wurden anxiolytische Effekte von Aripiprazol beschrieben [7]. Als Mechanismus wurden besonders die Effekte von Aripiprazol an zentralen 5-HT1A- und 5-HT2A-Rezeptoren diskutiert [4]. Da aber die Aripiprazol-Dosis bei diesen Untersuchungen im Mittel bei 10 mg pro Tag lag, erscheinen diese Mechanismen bei der relativ geringen Rezeptorokkupation unter dieser Dosis [30] allein nicht ausreichend. Sehr viel wahrscheinlicher ist die Annahme, dass die bei dieser Dosierung sehr deutliche Bindung an präsynaptische D2-Rezeptoren und die damit verbundene Aktivierung der dopaminergen Neurotransmission den Hauptmechanismus der Wirkung von Aripiprazol bei affektiven Syndromen darstellt. Dies geht parallel mit Befunden, dass die Wirkung von Aripiprazol in einem Angstmodell an der Maus 5-HT1A-unabhängige Effekte zeigt [14]. Die präferenzielle Blockade bestimmter präsynaptischer D2-Rezeptoren kann durch unterschiedliche Rezeptorreserven (siehe Abschnitt „Antipsychotischer Wirkungsmechanismus“) in einigen Strukturen, die relevant für die Affektmodulation sind, erklärt werden.
Aripiprazol hat in verschiedenen Studien an schizophrenen Patienten Verbesserungen der kognitiven Einschränkungen gezeigt [2, 19, 25, 31, 47, 48]. Auch an der Ratte wurden Kognitions-verbessernde Eigenschaften von Aripiprazol beschrieben [7]. Solche Effekte sind keine Gruppeneigenschaft der Antipsychotika allgemein, sondern nur für einige andere, aber auch nicht alle der atypischen Substanzen bekannt [27]. Gemeinsamer Mechanismus scheint eine Erhöhung der präfrontalen Dopamin-Konzentration zu sein [28], wie sie auch für die anderen Kognitions-verbessernden Atypika beschrieben wurde [27]. Auf neuronaler Ebene scheint ein partialagonistischer Effekt an 5-HT1A-Rezeptoren im Vordergrund zu stehen [54], die möglicherweise auch aktivierende Effekte der Neuroplastizität auslösen [50]. Wie erwähnt, wurden die zusätzlichen Effekte von Aripiprazol auf Affekt und Kognition bereits bei der Akutbehandlung der Schizophrenie als günstig diskutiert und könnten daher auch bei der Langzeitbehandlung mit Aripiprazol-Depot von Vorteil sein.
Zusammenfassung
Eine ganze Reihe von günstigen pharmakokinetischen und pharmakologischen Eigenschaften erklärt die bisher sehr guten klinischen Daten über Aripiprazol-Depot in der Erhaltungstherapie der Schizophrenie. Neben der langsamen Kinetik des Depots und der langen terminalen Eliminationshalbwertszeit sind es auf pharmakologischer Seite der partielle Agonismus am D2-Rezeptor und die zusätzliche Bindung an 5-HT2A- und 5-HT1A-Rezeptoren, die das sehr günstige Nebenwirkungsprofil bei guter antipsychotischer Wirkung erklären.
Interessenkonflikterklärung
Der Autor war in den letzten Jahren als wissenschaftlicher Berater und/oder Vortragender für folgende Firmen tätig: Astra-Zeneca, Cassella-med, Janssen, Lundbeck, Schwabe.
Literatur
1. Aihara K, Shimada J, Miwa T, Tottori K, et al. The novel antipsychotic aripiprazole is a partial agonist at short and long isoforms of D2-receptors linked to the regulation of adenylyl cyclase activity and prolactin release. Brain Res 2004;1003:9–17.
2. Bervoets C1, Morrens M, Vansteelandt K, Kok F, et al. Effect of aripiprazole on verbal memory and fluency in schizophrenic patients: results from the ESCAPE study. CNS Drugs 2012;26:975–82.
3. Bitter I, Katona L, Zámbori J, Takács P, et al. Comparative effectiveness of depot and oral second generation antipsychotic drugs in schizophrenia: a nationwide study in Hungary. Eur Neuropsychopharmacol 2013;23:1383–90.
4. Blier P, Blondeau C. Neurobiological bases and clinical aspects of the use of aripiprazole in treatment-resistant major depressive disorder. J Affect Disord 2011;128(Suppl 1):S3–10.
5. Bortolozzi A1, Díaz-Mataix L, Toth M, Celada P, et al. In vivo actions of aripiprazole on serotonergic and dopaminergic systems in rodent brain. Psychopharmacology (Berl) 2007;191:745–58.
6. Boulton DW, Kollia G, Mallikaarjun S, et al. Pharmacokinetics and tolerability of intramuscular, oral and intravenous aripiprazole in healthy subjects and in patients with schizophrenia. Clin Pharmacokinet 2008;47:475–85.
7. Burda K, Czubak A, Kus K, Nowakowska E, et al. Influence of aripiprazole on the antidepressant, anxiolytic and cognitive functions of rats. Pharmacol Rep 2011;63:898–907.
8. Burris KD, Molski TF, Xu C, Ryan E, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2-receptors. J Pharmacol Exp Ther 2002;302:381–9.
9. Caccia S. Pharmacokinetics and metabolism update for some recent antipsychotics. Expert Opin Drug Metab Toxicol 2011;7:829–46.
10. Citrome L. New second-generation long-acting injectable antipsychotics for the treatment of schizophrenia. Expert Rev Neurother 2013;13:767–83.
11. De Berardis D, Marini S, Carano A, Lang AP, et al. Efficacy and safety of long acting injectable atypical antipsychotics: a review. Curr Clin Pharmacol 2013;8:256–64.
12. De Leon A, Patel NC, Crismon ML. Aripiprazole: a comprehensive review of its pharmacology, clinical efficacy, and tolerability. Clin Ther 2004;26:649–66.
13. Dhillon S. Aripiprazole: a review of its use in the management of mania in adults with bipolar I disorder. Drugs 2012;72:133–62.
14. Egashira N, Okuno R, Matsushita M, Abe M, et al. Aripiprazole inhibits marble-burying behavior via 5-hydroxytryptamine(5-HT)1A-receptor-independent mechanisms. Eur J Pharmacol 2008;592:103–8.
15. Fachinformation Abilify® (2013).
16. Fachinformation Abilify Maintena® (2013).
17. Fleischhacker WW. Second-generation antipsychotic long-acting injections: systematic review. Br J Psychiatry Suppl 2009;52:S29–36.
18. Gentile S. Adverse effects associated with second-generation antipsychotic long-acting injection treatment: a comprehensive systematic review. Pharmacotherapy 2013;33:1087–106.
19. Hori H, Yoshimura R, Katsuki A, Hayashi K, et al. The cognitive profile of aripiprazole differs from that of other atypical antipsychotics in schizophrenia patients. J Psychiatr Res 2012;46:757–61.
20. Howes OD1, Kambeitz J, Kim E, Stahl D, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry 2012;69:776–86.
21. Jordan S, Koprivica V, Chen R, Tottori K, et al. The antipsychotic aripiprazole is a potent, partial agonist at the human 5-HT1A-receptor. Eur J Pharmacol 2002;441:137–40.
22. Kane JM1, Sanchez R, Perry PP, Jin N, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry 2012;73:617–24.
23. Kasper S, Lerman MN, McQuade RD, Saha A, et al. Efficacy and safety of aripiprazole vs. haloperidol for long-term maintenance treatment following acute relapse of schizophrenia. Int J Neuropsychopharmacol 2003;6:325–37.
24. Katzman MA. Aripiprazole: a clinical review of its use for the treatment of anxiety disorders and anxiety as a comorbidity in mental illness. J Affect Disord 2011;128(Suppl 1):S11–20.
25. Kern RS, Green MF, Cornblatt BA, Owen JR, et al. The neurocognitive effects of aripiprazole: an open-label comparison with olanzapine. Psychopharmacology (Berl) 2006;187:312–20.
26. Kinghorn WA, McEvoy JP. Aripiprazole: pharmacology, efficacy, safety and tolerability. Expert Rev Neurother 2005;5:297–307.
27. Leuner K, Müller WE. Dopamine in the prefrontal cortex and its different modulation by conventional and atypical antipsychotics. Pharmacopsychiatry 2007;40 (Suppl 1):S17–26.
28. Li Z, Ichikawa J, Dai J, Meltzer HY. Aripiprazole, a novel antipsychotic drug, preferentially increases dopamine release in the prefrontal cortex and hippocampus in rat brain. Eur J Pharmacol 2004;493:75–83.
29. Mallikaarjun S1, Kane JM, Bricmont P, et al. Pharmacokinetics, tolerability and safety of aripiprazole once-monthly in adult schizophrenia: an open-label, parallel-arm, multiple-dose study. Schizophr Res 2013;150:281–8.
30. Mamo D, Graff A, Mizrahi R, Shammi CM, et al. Differential effects of aripiprazole on D2-, 5-HT2-, and 5-HT1A-receptor occupancy in patients with schizophrenia: a triple tracer PET study. Am J Psychiatry 2007;164:1411–7.
31. Maat A, Cahn W, Gijsman HJ, Hovens JE, et al. Open, randomized trial of the effects of aripiprazole versus risperidone on social cognition in schizophrenia. Eur Neuropsychopharmacol 2014;24:575–84.
32. Mauri MC, Volonteri LS, Colasanti A, Fiorentini A, et al. Clinical pharmacokinetics of atypical antipsychotics: a critical review of the relationship between plasma concentrations and clinical response. Clin Pharmacokinet 2007;46:359–88.
33. McIntyre RS, Soczynska JK, Woldeyohannes HO, Miranda A, et al. Aripiprazole: pharmacology and evidence in bipolar disorder. Expert Opin Pharmacother 2007;8:1001–9.
34. McKeage K. Aripiprazole: a review of its use in the treatment of manic episodes in adolescents with bipolar I disorder. CNS Drugs 2014;28:171–83.
35. Molden E, Lunde H, Lunder N, Refsum H. Pharmacokinetic variability of aripiprazole and the active metabolite dehydroaripiprazole in psychiatric patients. Ther Drug Monit 2006;28:744–9.
36. Motiwala FB, Siscoe KS, El-Mallakh RS. Review of depot aripiprazole for schizophrenia. Patient Prefer Adherence 2013;7:1181–7.
37. Müller WE, Eckert A. Psychopharmakotherapie: pharmakologische Grundlagen. In: Möller HJ, Laux G, Kampfhammer HP (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Band 1: Allgemeine Psychiatrie. Heidelberg: Springer, 2011.
38. Müller WE. Partieller D2-Agonismus und dopaminerge Stabilisierung durch Aripiprazol. Psychopharmakotherapie 2002;9:120–7.
39. Park MH, Han C, Pae CU, Lee SJ, et al. Aripiprazole treatment for patients with schizophrenia: from acute treatment to maintenance treatment. Expert Rev Neurother 2011;11:1541–52.
40. Park EJ, Amatya S, Kim MS, Park JH, et al. Long-acting injectable formulations of antipsychotic drugs for the treatment of schizophrenia. Arch Pharm Res 2013;36:651–9.
41. Pigott TA, Carson WH, Saha AR, Torbeyns AF, et al.; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry 2003;64:1048–56.
42. Pogarell O1, Koch W, Karch S, Dehning S, et al. Dopaminergic neurotransmission in patients with schizophrenia in relation to positive and negative symptoms. Pharmacopsychiatry 2012;45(Suppl 1):S36–41.
43. Potkin SG, Raoufinia A, Mallikaarjun S, Bricmont P, et al. Safety and tolerability of once monthly aripiprazole treatment initiation in adults with schizophrenia stabilized on selected atypical oral antipsychotics other than aripiprazole. Curr Med Res Opin 2013;29:1241–51.
44. Prescribing information Abilify Maintena. USA. Otsuka Pharmceutica Co. 2013.
45. Rauch AS, Fleischhacker WW. Long-acting injectable formulations of new-generation antipsychotics: a review from a clinical perspective. CNS Drugs 2013;27:637–52.
46. Rummel-Kluge C, Komossa K, Schwarz S, Hunger H, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res 2010;123:225–33.
47. Sato G, Yoshimura S, Yamashita H, et al. The neurocognitive effects of aripiprazole compared with risperidone in the treatment of schizophrenia. Hiroshima J Med Sci 2012;61:75–83.
48. Schlagenhauf F, Dinges M, Beck A, Wüstenberg T, et al. Switching schizophrenia patients from typical neuroleptics to aripiprazole: effects on working memory dependent functional activation. Schizophr Res. 2010;118:189–200.
49. Schotte A, Bonaventure P, Janssen PF, Leysen JE. In vitro receptor binding and in vivo receptor occupancy in rat and guinea pig brain: risperidone compared with antipsychotics hitherto used. Jpn J Pharmacol 1995;69:399–412.
50. Schreiber R, Newman-Tancredi A. Improving cognition in schizophrenia with antipsychotics that elicit neurogenesis through 5-HT1A-receptor activation. Neurobiol Learn Mem 2014;110C:72–80.
51. Shapiro DA, Renock S, Arrington E, Chiodo LA, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400–11.
52. Sparshatt A, Taylor D, Patel MX, Kapur S. A systematic review of aripiprazole – dose, plasma concentration, receptor occupancy, and response: implications for therapeutic drug monitoring. J Clin Psychiatry 2010;71:1447–56.
53. Stark AD, Jordan S, Allers KA, Bertekap RL, et al. Interaction of the novel antipsychotic aripiprazole with 5-HT1A- and 5-HT2A-receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacology (Berl) 2007;190:373–82.
54. Sumiyoshi T, Higuchi Y, Uehara T. Neural basis for the ability of atypical antipsychotic drugs to improve cognition in schizophrenia. Front Behav Neurosci 2013;7:140.
55. Tadori Y, Miwa T, Tottori K, Burris KD, et al. Aripiprazole’s low intrinsic activities at human dopamine D2L and D2S receptors render it a unique antipsychotic. Eur J Pharmacol 2005;515:10–9.
56. Tadori Y, Forbes RA, McQuade RD, Kikuchi T. Receptor reserve-dependent properties of antipsychotics at human dopamine D2-receptors. Eur J Pharmacol 2009;607:35–40.
57. Tadori Y, Forbes RA, McQuade RD, Kikuchi T. In vitro pharmacology of aripiprazole, its metabolite and experimental dopamine partial agonists at human dopamine D2 and D3-receptors. Eur J Pharmacol 2011;668:355–65.
58. Takeuchi H, Remington G. A systematic review of reported cases involving psychotic symptoms worsened by aripiprazole in schizophrenia or schizoaffective disorder. Psychopharmacology (Berl) 2013;228:175–85.
59. Tourian L, Margolese HC. Extended-release intramuscular aripiprazole for maintenance pharmacotherapy in schizophrenia and related disorders. Neuropsychiatry 2013;3:345–54.
60. Wood MD, Scott C, Clarke K, Westaway J, et al. Aripiprazole and its human metabolite are partial agonists at the human dopamine D2-receptor, but the rodent metabolite displays antagonist properties. Eur J Pharmacol 2006;546:88–94.
61. Wood M, Reavill C. Aripiprazole acts as a selective dopamine D2-receptor partial agonist. Expert Opin Investig Drugs 2007;16:771–5.
62. Yokoi F, Gründer G, Biziere K, Stephane M, et al. Dopamine D2 and D3-receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology 2002;27:248–59.
63. Zhornitsky S, Stip E. Oral versus long-acting injectable antipsychotics in the treatment of schizophrenia and special populations at risk for treatment non-adherence: A systematic review. Schizophr Res Treatment 2012;2012:407171. doi: 10.1155/2012/407171. Epub 2012 Feb 15.
Univ.-Prof. Dr. Walter E. Müller, Höhenstraße 49A, 67550 Worms, E-Mail: w.e.mueller@em.uni-frankfurt.de
Therapeutic application of aripiprazole-depot: pharmacologic and pharmacokinetic basic principles
Aripiprazole-depot became recently available for the maintenance treatment of schizophrenia. After intramuscular injection of the microparticles made by lyophilisation, aripiprazole is only slowly released into the blood allowing a dosing interval of four weeks with a fairly stable blood level due to the long dissolution half-life of the depot and the long elimination half-life of plasma aripiprazole. The atypical properties of aripiprazole including good antipychotic activity and low side effect profile with little EPS, no weight gain or metabolic changes and no sedation are mainly explained by its property as a dopamine receptor partial agonist. It is not sedative and seems to have rather cognition enhancing than cognition reducing properties.
Key words: Aripiprazole depot, pharmacokinetics, partial dopamine recepor agonism, tolerability
Psychopharmakotherapie 2014; 21(03)