J. Farzan, Langen
Erweiterter Nebenwirkungsbegriff umfasst den nicht bestimmungsgemäßen Gebrauch
Die Erweiterung des Nebenwirkungsbegriffs stellt eine grundlegende Änderung dar. Zukünftig müssen Zulassungsinhaber den zuständigen Bundesoberbehörden BfArM und PEI sämtliche schädlichen und unbeabsichtigten Reaktionen auf Humanarzneimittel melden, auch wenn sie durch nicht bestimmungsgemäßen Gebrauch entstanden sind. Dies betrifft insbesondere auch Off-Label-Use und Medikationsfehler. Der bisherige Nebenwirkungsbegriff bezog sich lediglich auf den bestimmungsgemäßen Gebrauch, also die Anwendung entsprechend der Fach- und Gebrauchsinformation. Auch Ärzte, Apotheker und Patienten sind aufgerufen, schädliche Reaktionen durch nicht bestimmungsgemäßen Gebrauch anzuzeigen. Hierdurch sollen die Arzneimittelsicherheit zusätzlich gestärkt und die Anwendung des Produkts und die damit verbundenen Risiken nunmehr in ihrer Gesamtheit erfasst werden.
Meldung durch Patienten
Die Richtlinie sieht ferner vor, dass ab Juli 2012 nicht nur Angehörige der Gesundheitsberufe, sondern auch Patienten Verdachtsfälle von Nebenwirkungen den Zulassungsbehörden melden sollen. Darin zeigt sich ein verändertes Verständnis des informierten und kritischen Verbrauchers, der nunmehr auf die gleiche Ebene wie die Fachkreise gehoben wird und sich aktiv am Spontanerfassungssystem beteiligen soll. In den Packungsbeilagen wird daher ein Standardtext aufgenommen, wonach die Patienten ausdrücklich aufgefordert werden, jeden Fall einer Nebenwirkung ihren Ärzten, Apothekern oder unmittelbar den zuständigen Bundesoberbehörden BfArM und PEI zu melden. Die Meldung kann in jeder Form erfolgen, insbesondere auch elektronisch. In Deutschland führen die neuen Bestimmungen der Richtlinie indes zu wenig Änderungen in der Praxis, da Patientenmeldungen auch bislang bereits registriert und bewertet wurden.
Meldung nicht schwerwiegender Nebenwirkungen
War der Zulassungsinhaber bisher nur verpflichtet, ihm bekannte Verdachtsfälle schwerwiegender Nebenwirkungen als Einzelfallbericht innerhalb von 15 Tagen den Zulassungsbehörden zu melden, so hat er zukünftig auch Verdachtsfälle nicht schwerwiegender Nebenwirkungen als Einzelfallbericht innerhalb von 90 Tagen anzuzeigen. Bislang wurden nicht schwerwiegende Meldungen lediglich im Rahmen der regelmäßig aktualisierten Unbedenklichkeitsberichte summarisch dargestellt und bewertet.
Arzneimittel unter zusätzlicher Überwachung
Eine weitere Änderung betrifft ebenfalls die Packungsbeilage: So wird die EMA eine Liste mit Humanarzneimitteln veröffentlichen, die zusätzlicher Überwachung bedürfen. Betroffen hiervon sind Produkte mit neuen, seit dem 1. Januar 2012 zugelassenen Wirkstoffen, insbesondere auch Biologika. Diese Arzneimittel sind weitergehenden Pharmakovigilanzmaßnahmen wie etwa Unbedenklichkeitsstudien nach der Zulassung unterworfen, um die Sicherheit dieser Arzneimittel zusätzlich zu untersuchen. Eine entsprechende Kennzeichnung durch ein schwarzes Symbol erfolgt auf der Fachinformation und der Packungsbeilage.
Internetportal für Arzneimittel
Zur Verbesserung der Transparenz und Information soll bei der EMA und in allen europäischen Mitgliedstaaten bis Juli 2012 jeweils ein Internetportal zur Arzneimittelsicherheit mit bestimmten, vereinheitlichten Inhalten geschaffen werden. Auf dem europäischen Portal finden sich dann unter anderem Informationen für Patienten, Ärzte und Apotheker darüber, wie man den nationalen Zulassungsbehörden Verdachtsfälle von Nebenwirkungen meldet, einschließlich entsprechender Meldeformulare. Ferner ist eine ständig aktualisierte Liste mit den von der EMA zentralisiert zugelassenen Arzneimitteln enthalten, die zusätzlicher Überwachung unterworfen sind. Von hohem Nutzen für die Transparenz ist auch, dass zukünftig sämtliche Entscheidungen, Gutachten und Stellungnahmen der europäischen Ausschüsse und Gremien einsehbar sind. Hier kann jedermann, also Patienten ebenso wie Ärzte, Apotheker und Fachkreise, Einblick in Verfahren und Entscheidungsprozesse nehmen und hieran auch aktiv – etwa im Rahmen von öffentlichen Anhörungen – teilnehmen. Die beiden Bundesoberbehörden werden die EMA-Homepage auf ihrer Homepage verlinken.
Neuer Ausschuss für Risikobewertung der EMA
Durch die Änderung der Richtlinie 2001/83/EG werden zahlreiche Entscheidungskompetenzen auch für national zugelassene Humanarzneimittel von den nationalen Zulassungsbehörden auf die EMA und dessen neu geschaffenen Ausschuss für Risikobewertung (Pharmacovigilance Risk Assessment Committee, PRAC) übertragen. Neben den Vertretern der Mitgliedstaaten und der Kommission gehören dem Ausschuss auch jeweils ein Vertreter der Angehörigen der Gesundheitsberufe und der Patienten an. Der PRAC löst die bisherige Pharmakovigilanz-Arbeitsgruppe der EMA ab.
Bewertung von nationalen Zulassungen durch die EMA
Der PRAC nimmt zukünftig eine zentrale Stellung in der Pharmakovigilanz ein, und zwar sowohl für von der EMA zentralisiert zugelassene Produkte wie auch für solche, die im Verfahren der gegenseitigen Anerkennung, im dezentralisierten Verfahren oder rein national in mehreren europäischen Mitgliedstaaten zugelassen sind. Das Mandat des Ausschusses umfasst alle Aspekte des Risikomanagements, also die Einleitung von Maßnahmen zur Ermittlung, Beschreibung, Vermeidung oder Minimierung von Arzneimittelrisiken.
Für die Signalerkennung und Bewertung der Nebenwirkungsmeldungen werden die nationalen Behörden BfArM und PEI in Zusammenarbeit mit dem PRAC zuständig sein. Welche Maßnahmen dann zur Risikominimierung getroffen werden sollen, wird zukünftig indes auch bei nationalen Zulassungen, sofern das Arzneimittel in mehr als einem Mitgliedsland zugelassen ist, nicht mehr auf nationaler, sondern auf europäischer Ebene entschieden.
Dringlichkeitsverfahren
Soweit aufgrund der Bewertung von Daten aus der Pharmakovigilanz – wie etwa der Auswertung von Nebenwirkungsmeldungen – dringendes Handeln als notwendig erachtet wird, können BfArM und PEI oder die Behörden der anderen Mitgliedstaaten ebenso wie die Europäische Kommission ein Verfahren einleiten, um die betroffene Zulassung gegebenenfalls zu widerrufen oder deren Ruhen anzuordnen. Dieses europäische Verfahren wird auch bei nationalen Zulassungen ausgelöst, soweit diese in mehr als einem Mitgliedstaat existieren. BfArM und PEI können allenfalls vorübergehend bei Gefahr im Verzug nationale Sofortmaßnahmen einleiten; die endgültige Bewertung erfolgt durch den PRAC.
Das Verfahren gestaltet sich im Einzelnen wie folgt: Handelt es sich um eine zentralisierte Zulassung der Europäischen Kommission, so erstellt der Ausschuss für Humanarzneimittel der EMA (CHMP) auf Grundlage der Empfehlung des PRAC ein Gutachten, welches wiederum der Kommission zwecks endgültiger Entscheidung weitergeleitet wird. Geht es hingegen um eine nationale Zulassung beziehungsweise eine im Verfahren der gegenseitigen Anerkennung oder im dezentralisierten Verfahren erteilte Zulassung, so formuliert die Koordinierungsgruppe der Mitgliedstaaten einen Standpunkt hinsichtlich der zu treffenden Entscheidung, den die Mitgliedstaaten dann umsetzen müssen. Fällt der Standpunkt der Koordinierungsgruppe nicht einstimmig aus, so trifft die Europäische Kommission die Entscheidung.
Die EMA gibt die Einleitung des Dringlichkeitsverfahrens über das europäische Internetportal für Arzneimittel öffentlich bekannt. Hierbei ergibt sich die Möglichkeit für die Angehörigen der Gesundheitsberufe ebenso wie für die übrige Öffentlichkeit, der EMA verfahrensrelevante Informationen zu übermitteln. Darüber hinaus kann der PRAC eine öffentliche Anhörung durchführen, soweit die Dringlichkeit es zulässt. Wie diese öffentliche Anhörung durchgeführt wird und auf welche Weise Ärzte, Apotheker und Patienten hieran teilnehmen können, wird die EMA zuvor ebenfalls auf ihrem Internetportal ankündigen. In jedem Fall wird diese Verfahrensweise zu mehr Information und Transparenz für die Öffentlichkeit führen, und Patienten wie Fachkreise werden dazu ermutigt, sich aktiv am Verfahren zu beteiligen.
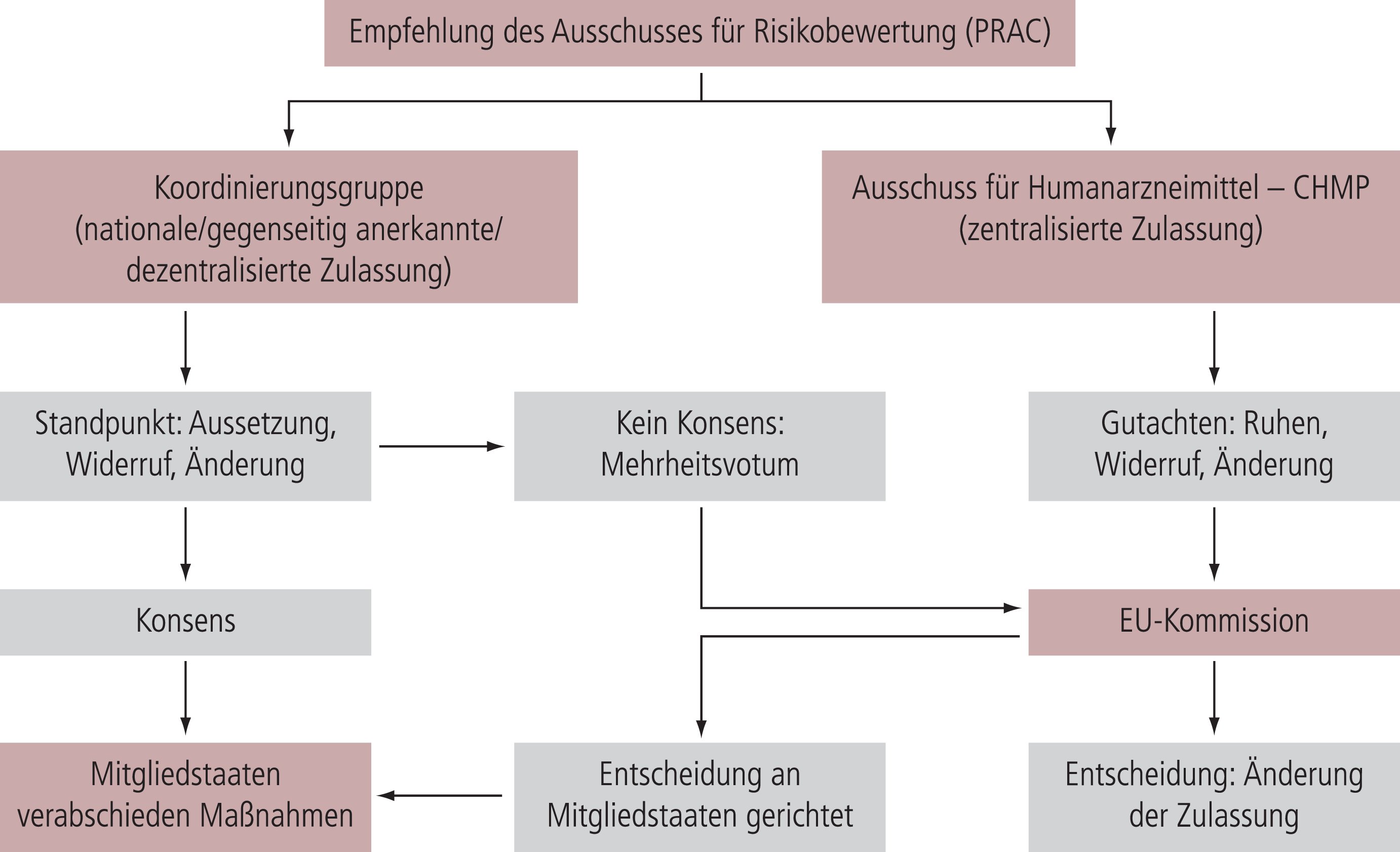
Abb. Entscheidungsverfahren der EU. Dringlichkeitsverfahren der EU, um aus Pharmakovigilanz-Gründen die Zulassung zu widerrufen, auszusetzen oder zu ändern.
Risikomanagement-System
Damit es gar nicht erst zu dem dargestellten Verfahren kommt, setzt das neue, von der EU propagierte Konzept auf proaktive Pharmakovigilanz. Um etwaige Arzneimittelrisiken rasch erkennen und minimieren zu können, wird die Einführung eines Risikomanagement-Systems für neu zugelassene Arzneimittel verbindlich. Dies ist in der Praxis zwar bereits jetzt fester Bestandteil neuer Zulassungen, wird nunmehr aber explizit gesetzlich festgeschrieben. Für bereits zugelassene Arzneimittel kann ebenfalls ein Risikomanagement-System von den Behörden gefordert werden. Das Risikomanagement-System beinhaltet eine Reihe von Maßnahmen, durch die Risiken im Zusammenhang mit Arzneimitteln ermittelt, beschrieben, vermieden oder minimiert werden sollen.
Nichtinterventionelle Unbedenklichkeitsstudien
Eine weitere wichtige Änderung mit Relevanz für betroffene Ärzte stellen die neuen Bestimmungen zu nichtinterventionellen Unbedenklichkeitsstudien nach der Zulassung dar. So hat der Zulassungsinhaber Studienprotokolle zukünftig vor Studienbeginn dem PRAC anzuzeigen, soweit die Studie in mehreren Mitgliedstaaten betrieben wird. Deren Durchführung wird versagt, wenn durch sie die Anwendung eines Arzneimittels gefördert werden soll, Vergütungen für teilnehmende Ärzte nicht auf den tatsächlichen Kosten- und Zeitaufwand beschränkt sind oder ein Anreiz für eine bevorzugte Verschreibung oder Empfehlung bestimmter Produkte entsteht.
Neu ist auch, dass BfArM und PEI die Zulassungsinhaber durch Erteilung entsprechender Auflagen zur Durchführung von Unbedenklichkeitsstudien verpflichten können. Bei den behördlich auferlegten Studien erfolgen die Genehmigung des Protokolls und die Bewertung der Studienergebnisse ebenfalls durch den PRAC, soweit die Studie in mehreren Ländern stattfindet.
Wirksamkeitsstudien nach der Zulassung
Erwähnt sei zu guter Letzt die Einführung von Wirksamkeitsstudien nach der Zulassung. Dem Zulassungsinhaber kann zukünftig bei Zulassungserteilung die nachträgliche Durchführung derartiger Studien auferlegt werden, soweit Fragen zur Wirksamkeit bestehen und diese erst nach Inverkehrbringen geklärt werden können. Nach Zulassung kann eine Wirksamkeitsstudie gefordert werden, wenn neue Erkenntnisse über die Krankheit oder die klinische Methodik auf die Notwendigkeit einer Korrektur der bisherigen Wirksamkeitsbewertung hindeuten.
Fazit
Die Richtlinie 2001/83/EG führt zu einer europaweiten Harmonisierung und Zentralisierung der Pharmakovigilanz. Insbesondere bei nationalen Zulassungen verlagern sich zukünftig die Entscheidungskompetenzen von den nationalen Zulassungsbehörden auf die EMA und dessen Ausschuss für Risikobewertung PRAC, auch wenn natürlich der PRAC die nationale Expertise mit umfasst. Zugleich bezweckt die Richtlinie, Transparenz und Information für Verbraucher und Fachkreise in der gesamten Europäischen Union zu verbessern.
*Nachdruck aus Bulletin für Arzneimittelsicherheit 2011;(3):14–7 mit freundlicher Genehmigung.
Jan Farzan, Justiziar, Paul-Ehrlich-Institut, Paul-Ehrlich-Straße 51–59, 63225 Langen
Psychopharmakotherapie 2011; 18(06)