Gabriele Blaeser-Kiel, Hamburg
Die ubiquitär im Körper vorkommenden Kaliumkanäle unterscheiden sich in ihrem Aufbau, den Aktivierungsmechanismen und der gewebespezifischen Expression. Über hundert verschiedene Gene kodieren für unterschiedliche Untereinheiten. Ein für die Epileptologie interessantes Target sind spannungsabhängige Kaliumkanäle der KCNQ-Genfamilie (Kv7-Kanäle). Weil sich diese Kanäle experimentell mit Muscarin blockieren lassen, wird der von ihnen generierte Kaliumstrom als M-Strom bezeichnet.
Relativ selektiv im zentralen und peripheren Nervensystem werden die Subtypen Kv7.2 und Kv7.3 exprimiert. In Neuronen sind diese Kaliumkanäle am Axoninitialsegment in unmittelbarer Nachbarschaft von Natriumkanälen lokalisiert und können dadurch das Membranpotenzial im Subschwellenbereich eines Aktionspotenzials regulieren. Eine Aktivierung der Kanäle führt zu einer Hyperpolarisation und einer geringeren Erregbarkeit der Zelle, eine Blockade der Kanäle dagegen zur neuronalen Übererregbarkeit.
Kv7-Kanäle
Die KCNQ-Gene kodieren für insgesamt fünf verschiedene Untereinheiten der Kv7-Kanäle (KCNQ-Kanäle). Die Untereinheiten Kv7.2, Kv7.3 und Kv7.5 sind vorwiegend im Gehirn und in den Ganglien anzutreffen, die Untereinheit Kv7.1 vorwiegend am Herzen und wie die Untereinheit Kv7.4 im Innenohr.
Kaliumkanäle bestehen aus jeweils vier Untereinheiten, die gleichartig (homomer) oder, wie bei Kv7.2/7.3-Kanälen, verschieden (heteromer) sein können. (Red.)
Erstmals in das Blickfeld der Epileptologen gelangten die Kv7.2/7.3-Kanäle, als ihre Beteiligung bei der Pathogenese der sogenannten gutartigen familiären Neugeborenenkrämpfe (benign familial neonatal convulsions, BFNC) entdeckt wurde. Mutationen in den Genen KCNQ2 und KCNQ3 bewirken einen Ausfall oder eine Aktivitätsminderung der Kanäle, die zu einer erhöhten Feuerfrequenz der Neuronen führt. Ob der bei diesem Epilepsiesyndrom ursächlich beteiligte Defekt der Kaliumkanäle generell bei der Entstehung von Epilepsien eine Rolle spielt, kann heute noch nicht abschließend beurteilt werden. Es gibt jedoch Hinweise, dass die durch eine Fehlfunktion von Kaliumkanälen gesteigerte Erregbarkeit von Nervenzellen ein eher grundsätzlicher Pathomechanismus ist, dem sich mit einem Kaliumkanalöffner entgegenwirken lässt.
Retigabin
Der erste Vertreter dieses Wirkprinzips ist Retigabin (Abb. 1). Es bewirkt gewissermaßen das Gegenteil der bei Patienten mit benignen familiären Neigeborenenkrämpfen nachgewiesenen Mutationen. Retigabin bindet an Kaliumkanäle vom Typ Kv7.2 bis Kv7.5. In pharmakologischen Untersuchungen führte dies zur Stabilisierung des offenen Zustands dieser Kanäle und damit Steigerung ihrer Aktivität. Als Folge kam es zu einer Hyperpolarisation und einer verringerten Erregbarkeit der Zelle.
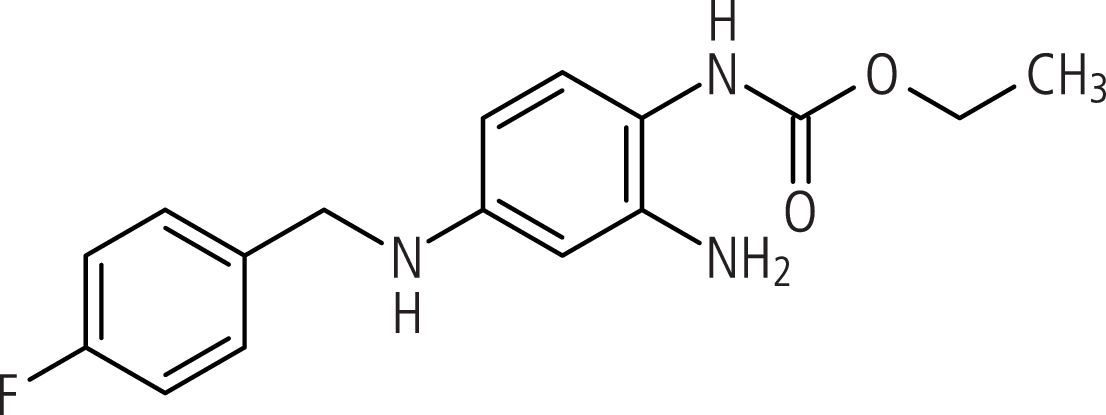
Abb. 1. Retigabin
Die Pharmakokinetik von Retigabin ist gekennzeichnet durch eine lineare Dosis-Wirkungs-Beziehung; nach oraler Applikation werden maximale Plasmakonzentrationen innerhalb von 1,5 bis 2 Stunden erreicht, die Halbwertszeit liegt bei 6 bis 10 Stunden. Die Verstoffwechslung zu inaktiven oder schwach aktiven Metaboliten erfolgt nicht über hepatische Cytochrom-P450-Enzyme, sondern hauptsächlich durch N-Glucuronidierung sowie N-Acetylierung. Dies lässt ein geringes Potenzial für Interaktionen mit anderen Antikonvulsiva und sonstiger Komedikation bei der Elimination erwarten.
Die Wirksamkeit und Sicherheit der neuen Substanz wurde unter anderem in zwei Phase-III-Studien mit den Akronym RESTORE (Retigabine efficacy and safety trial for partial onset epilepsy) untersucht [2, 3]. An den in Europa und den USA durchgeführten Studien nahmen Patienten mit therapierefraktären fokalen Epilepsien und hoher Krankheitsaktivität (im Median 9 bis 12 Anfälle pro Monat) teil. Durch den Zusatz von Retigabin zur bestehenden Therapie, die in mehr als 80% der Fälle aus Zwei- und Dreifachkombinationen bestand, wurde eine stärkere Anfallsreduktion erreicht als durch Zusatz von Plazebo. Die Add-on-Medikation führte zu Responseraten (mindestens 50%ige Anfallsreduktion) von 39% (600 mg/Tag), 47% (900 mg/Tag) und 56% (1200 mg/Tag) – p<0,01 bis p<0,001 versus Plazebo-Gabe (Responserate 19% bzw. 23%).
RESTORE-Studien
An den RESTORE-Studien nahmen insgesamt 843 Patienten mit fokaler Epilepsie teil, die trotz Behandlung mit 1 bis 3 etablierten Antiepileptika mindestens vier Anfälle pro 4 Wochen hatten. Randomisiert und doppelblind erhielten sie, verteilt auf drei Einzelgaben, Retigabin in einer Tagesdosis von 1200 mg (RESTORE 1), 900 mg oder 600 mg (RESTORE 2) oder Plazebo. Die Retigabin-Behandlung begann mit 300 mg/Tag und wurde in wöchentlichen 150-mg-Schritten bis zur Zieldosis erhöht; die Zieldosis wurde dann für 12 Wochen beibehalten.
Primärer Endpunkt war die Veränderung der 4-Wochen-Anfallsfrequenz (Median) zwischen Baseline und Doppelblindphase (Titrations- und Erhaltungsphase). Die Anfallsfrequenz sank mit 600 mg/Tag Retigabin um 27,9%, mit 900 mg/Tag um 39,9% und mit 1200 mg/Tag um 44,3% (Plazebo: –15,9% bzw. –17,5%).
Zwischen Baseline und Erhaltungsphase sank die 4-Wochen-Anfallsfrequenz im Median um 35,3% (600 mg), 44,3% (900 mg) bzw. 54,5% (1200 mg) (Plazebo: –17,4% bzw. –18,9%).
Eine mindestens 50%ige Reduktion der 4-Wochen-Anfallsfrequenz zwischen Baseline und Erhaltunsgphase erreichten 38,6% (600 mg), 47,0% (900 mg) und 55,5% (1200 mg) (Plazebo: 18,9% bzw. 22,6%) der Patienten.
Alle Ergebnisse waren hochsignifikant (meist p<0,001).
Studienabbrüche waren in beiden Studien unter Retigabin häufiger als unter Plazebo (14–27% vs. 8–9%).
Beide Studien werden als offene Studie fortgesetzt (open-label extension).
[Quellen: www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/ Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM221682.pdf (Zugriff 21.10.2010); www.clinicaltrials.gov] (Red.)
Die aufgetretenen Nebenwirkungen hingen vorwiegend mit der Dämpfung der neuronalen Erregbarkeit zusammen, wie Müdigkeit oder Schwindel. Weil Kanäle mit Kv7.3- und Kv7.5-Untereinheiten auch in der urogenitalen und gastrointestinalen Muskulatur exprimiert werden, wurde in den Studien sehr sorgfältig auf Beschwerden in diesen Bereichen geachtet. Dabei wurden Blasenentleerungstörungen aufgrund einer Beeinträchtigung der Detrusoraktivität bei rund 5% der Patienten gesehen (Plazebo: 3%). Kardiovaskuläre Komplikationen sind dagegen nach heutigem Kenntnisstand unwahrscheinlich, da Retigabin nicht an die im Herzen vorrangig exprimierte Untereinheit Kv7.1 bindet [1].
Quellen
1. Prof. Dr. med. Holger Lerche, Tübingen, Satellitensymposium „Schwer zu behandelnde Epilepsien – Definition und Therapie neu durchdacht“, veranstaltet von GlaxoSmithKline im Rahmen der 50. Jahrestagung der Deutschen Gesellschaft für Epileptologie, Wiesbaden, 29. April 2010.
2. French JA, et al. Retigabine 1200 mg/day as adjunctive therapy in adults with refractory partial-onset seizures. Poster 2405 beim 13th EFNS Congress 2009; Eur J Neurol 2009;16(Suppl 3):472.
3. Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology 2010; Epub ahead of print (DOI: 10.1212/WNL.0b013e3181fd6170)
Psychopharmakotherapie 2010; 17(06)