Wiebke Frank, Ulm, Alzbeta Mühlbäck, Ulm/Taufkirchen/Prag, Rainer Hoffmann, Taufkirchen, Katrin S. Lindenberg und Albert C. Ludolph, Ulm
Die Huntington-Krankheit (HK) ist eine monogenetische neurodegenerative Erkrankung, die sich meist im mittleren Lebensalter manifestiert und sich durch einen progressiven Verlauf mit einem breiten Spektrum an psychiatrischen Symptomen, zunehmenden kognitiven Einschränkungen sowie verschiedene Bewegungsstörungen (u. a. unwillkürliche Chorea, Dystonie, Okulomotorikstörungen, Dysarthrie, Dysphagie) auszeichnet [47]. Das Symptomprofil unterscheidet sich bei den einzelnen Patienten teils sehr deutlich und bedarf unterschiedlicher symptomatischer Therapien. Die Diagnose einer manifesten HK wird gestellt, wenn typische motorische Symptome auftreten [43]. Zuvor können sich bei Patienten in der Prodromalphase teilweise über mehrere Jahre subtile psychiatrische und kognitive Veränderungen zeigen [26]. In dieser Phase finden bereits neurobiologische Veränderungen statt, die mit einer Störung des kortiko-striatalen Netzwerks einhergehen und zu einer beginnenden striatalen Atrophie führen, die mittels bildgebender Verfahren, meist MRT, nachgewiesen werden kann [44]. Die Konversion von der prämanifesten und prodromalen Phase mit subtilen Verhaltensänderungen bis zur manifesten Erkrankung mit dem Auftreten von motorischen Symptomen, kann sich über mehrere Jahre hinziehen [25]. Eine kurative Therapie besteht bisher nicht. Daher konzentriert sich die Therapie mittels pharmakologischer und nichtpharmakologischer Ansätze auf eine symptomatische Behandlung zur Symptomlinderung. Die HK führt im Verlauf zu Pflegebedürftigkeit und nach einem durchschnittlichen Krankheitsverlauf von etwa 20 Jahren zum frühzeitigen Tod.
In der europäischen Bevölkerung ist gegenwärtig etwa einer von 10 000 Einwohnern von der HK betroffen [29]. Das bedeutet, dass in Deutschland bei 80 Millionen Einwohnern etwa 8000 Menschen mit HK leben [10].
Genetik der HK und pathogene Mechanismen
Die HK ist eine monogenetische, autosomal-dominante, neurodegenerative Erkrankung, die durch eine instabile Expansion des sich wiederholenden Basentripletts CAG (Cytosin-Adenin-Guanin) im Exon 1 des Huntingtin-Gens (HTT, zuvor als IT-15 beschrieben) auf Chromosom 4 verursacht wird [14]. Dabei besteht eine inverse Korrelation zwischen der Anzahl der CAG-Wiederholungen und dem Krankheitsbeginn bzw. -verlauf. Je mehr CAG-Wiederholungen vorliegen, desto früher treten die Symptome auf und desto schneller ist der Krankheitsprogress [21, 22]. Bei Vorliegen von 36 bis 39 CAG-Basentripletts im HTT-Gen besteht eine verminderte Penetranz und bei Auftreten von Krankheitssymptomen treten diese dann erst im höheren Lebensalter auf und sind weniger einschränkend [6]. Ab ≥ 40 CAG-Tripletts in einer Kopie des HTT-Gens kommt es zur Manifestation der HK innerhalb einer normalen Lebensdauer – meistens im mittleren Erwachsenenalter. In seltenen Fällen kann die CAG-Expansion außergewöhnlich lang sein, was bereits zu einem Krankheitsbeginn im Jugendalter oder Kindesalter führen kann (juvenile oder pädiatrische HK) [28]. Träger der Huntington-Mutation können durch eine molekulargenetische Untersuchung im Rahmen einer prädiktiven Testung mit zuvor erfolgter humangenetischer Beratung bereits vor dem Auftreten von Symptomen identifiziert werden.
Neben der genetischen Antizipation, das heißt der möglichen Zunahme der CAG-Wiederholungen bei Vererbung an die nachfolgende Generation, weist die instabile HTT-Mutation eine zusätzliche somatische Instabilität auf und es kommt im Laufe des Lebens zu einer deutlichen Verlängerung der CAG-Wiederholungen im HTT-Gen, insbesondere in Nervenzellen [15]. In genomweiten Assoziationsstudien (GWAS) konnten DNA-Reparaturgene als Modifier der HK und verschiedene DNA-Reparatur-Komponenten als ursächlich für die somatische Expansion der CAG-Wiederholungen im HTT-Gen [11, 21, 51] identifiziert werden (Abb. 1).
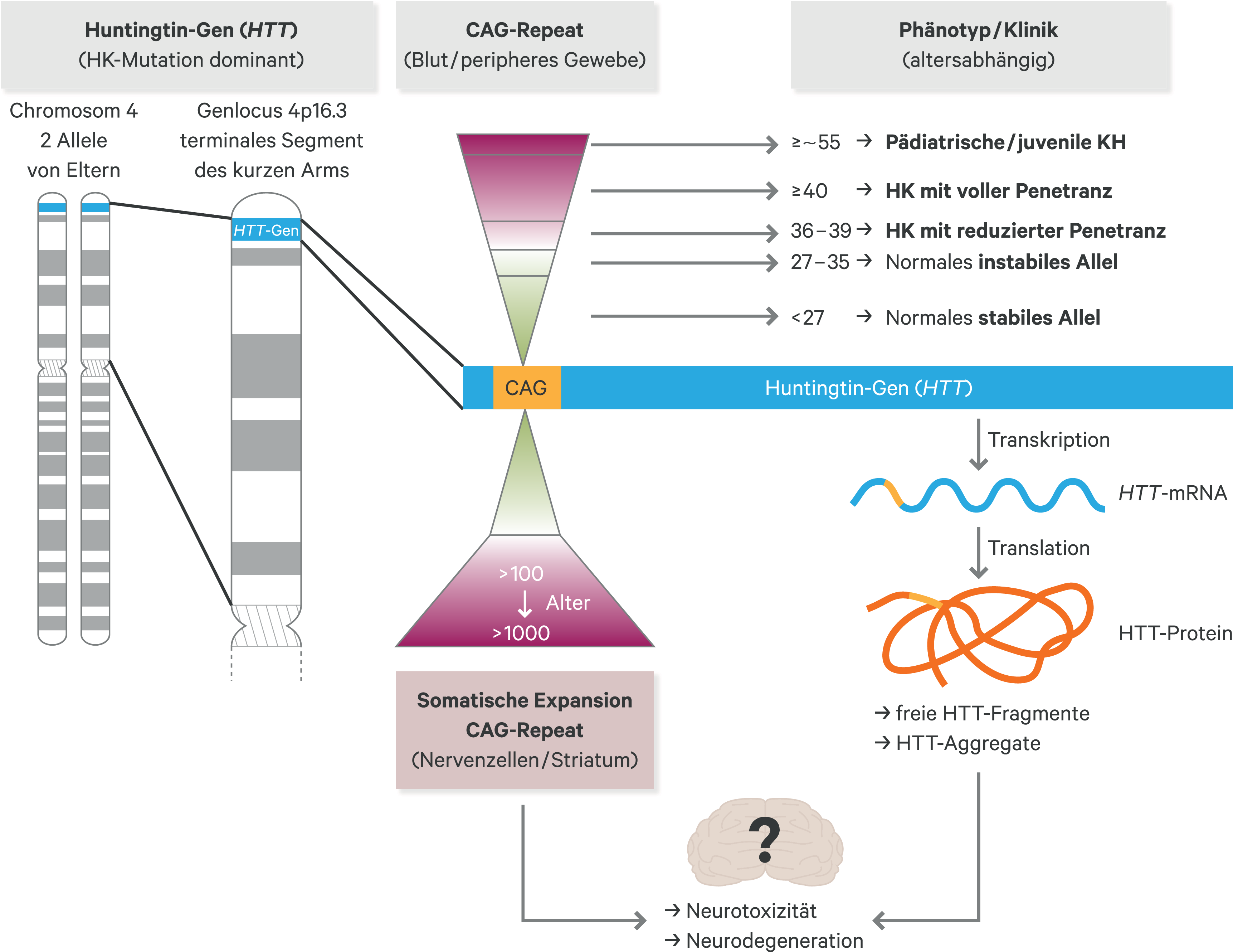
Abb. 1. Übersicht zur Genetik und Pathologie der Huntington-Krankheit Die Huntington-Mutation besteht aus einer krankheitsverursachenden Verlängerung im Exon-1 des HTT-Gens auf dem Chromosom 4, bei der das Basentriplett CAG pathologisch verlängert ist. Je höher die Anzahl der CAG-Wiederholungen ist, desto instabiler wird dieser Abschnitt der DNA. Bei CAG-Wiederholungen im HTT-Gen ≤ 27 ist dieser Sequenzabschnitt stabil. HK: Huntington-Krankheit, CAG: Cytosin-Adenin-Guanin, HTT: Huntingtin. Die expandierte mutante CAG-Wiederholung weist eine somatische Instabilität auf und verlängert sich über die Lebenszeit. Sehr lange CAG-Wiederholungen von > 100 bis vereinzelt > 1000 CAG-Repeats sind möglich (rotes Trapez).
Die pathologisch expandierte CAG-Wiederholungssequenz im HTT-Gen wird im Rahmen der Protein-Biosynthese in eine verlängerte Polyglutaminkette (polyQ) im Huntingtin-Protein (HTT) übersetzt [14]. Obwohl die genetische Ursache der HK seit 1993 bekannt ist, sind die molekularen und zellulären Veränderungen, die zu dem für die HK charakteristischen selektiven Neuronenverlust, insbesondere in den Basalganglien führen, noch nicht vollständig aufgeklärt. Zu den bisher identifizierten pathogenen Mechanismen zählen die Akkumulation und Aggregation von Amino-terminalen Fragmenten des mutanten HTT-Proteins [7]. Weiterhin wird ein Verlust der physiologischen Funktion des mutanten HTTs angenommen. Die physiologische Funktion des 346 kD großen HTTs, das im gesamten Organismus exprimiert wird, ist ebenfalls nur lückenhaft verstanden [5, 13]. Die Komplexität dieses Proteins ergibt sich aus den zahlreichen Interaktionsproteinen und verschiedenen post-translationalen Modifikationen [32]. Aufgrund der vielfältigen Funktionen des HTTs führt die Mutation zu Veränderungen der Transkription, des zellulären Energiehaushalts sowie aufgrund der Interaktion von HTT mit glutamatergen Rezeptoren zu Änderungen der neuronalen Transmission [49]. Neben der veränderten Proteinfunktion kommt es durch die mutierten HTT-mRNA-Transkripte zusätzlich zu Änderungen des Spleißvorgangs, die ebenfalls zur Pathogenese der HK beitragen [12]. Zusammenfassend tragen im Falle der HK auf DNA-, RNA- und auch Proteinebene unterschiedliche Pathomechanismen zur neuronalen Dysfunktion und schließlich zum Nervenzellverlust bei [4, 41]. Einige dieser pathobiologischen Mechanismen wurden im Laufe der Jahre bereits als Ansatzpunkte für therapeutische Interventionen herangezogen. Aktuell werden verschiedene neue Therapieansätze zur Behandlung der HK verfolgt, die vor allem auf die Verringerung der Neubildung von mutanten Genprodukten (z. B. HTT-mRNA, HTT-Protein) ausgerichtet sind. Eine Übersicht der zugrunde liegenden genetischen Ursache und Pathologie der HK ist in Abbildung 1 grafisch dargestellt.
Behandlungsansätze in der klinischen Entwicklung
Derzeit gibt es keine kurativen Therapien für die HK. Das verbesserte Verständnis der molekularen pathogenen Mechanismen hat jedoch maßgeblich zur Entwicklung von genspezifischen Behandlungsoptionen für die HK beigetragen. Diese innovativen Therapieansätze, die bereits in klinischen Studien untersucht werden, zielen darauf ab, die Bildung mutierter HTT-Genprodukte zu verringern. Es besteht die Hoffnung, durch den Einsatz der neuen Behandlungsmethoden das unaufhaltsame Fortschreiten der HK zu verlangsamen und – bei rechtzeitigem Behandlungsbeginn – präventiv den Krankheitsbeginn der HK hinauszuschieben oder im besten Fall verhindern zu können.
Es wird angenommen, dass das mutierte HTT-Protein durch einen überwiegend toxisch wirkenden „gain-of-function“-Mechanismus Zellschäden verursacht [1]. Zu den pathologischen zellulären Mechanismen gehören eine frühe transkriptionelle Dysregulation, synaptische Dysfunktion, extrasynaptische Exzitotoxizität, Beeinträchtigung der Proteostase, vermehrte Aggregatbildung, oxidativer Stress und die mitochondriale Dysfunktion [1, 7]. Bisher fokussierten sich die therapeutischen Strategien auf die gestörten zellulären Funktionen, wobei anfänglich vielversprechende Substanzen später in klinischen Studien keinen krankheitsmodifizierenden bzw. symptomatischen Effekt bei der HK zeigten [31, 45].
„Gene Silencing“ zur Reduktion der Neusynthese mutanter HTT-Genprodukte
Neue Therapieansätze sind auf die Hemmung der Genexpression ausgerichtet, die zur Reduktion des mHTT-Proteins beiträgt [50]. Eine der ersten Studien auf dem Gebiet der HK zeigte, dass die Verminderung der mhtt-Synthese im Striatum transgener Mäuse zu einer Verbesserung der Krankheitssymptome führte [48, 53]. Prinzipiell kann die Reduktion der Neusynthese von mHTT-Genprodukten entweder durch die Degradation von mRNA-Transkripten oder durch die Modulation der Transkription bereits auf DNA-Ebene erfolgen [50]. Diese möglichen Ansatzpunkte für eine therapeutische Intervention sind in Abbildung 2 grafisch dargestellt.
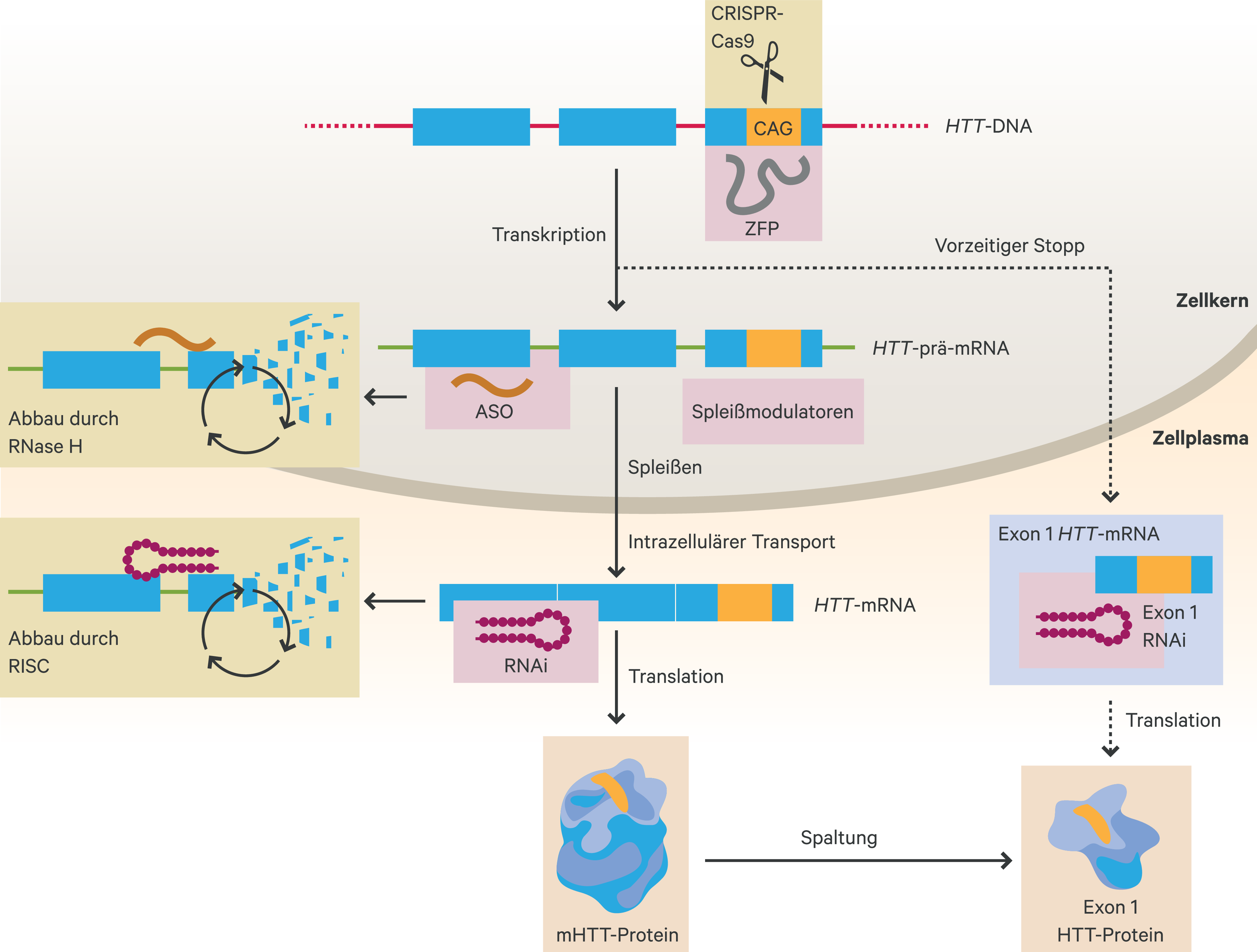
Abb. 2. Übersicht therapeutischer Ansatzpunkte für die Huntington-Krankheit (adaptiert nach [50]) ASO: Antisense-Oligonukleotid; RISC: RNA-induced silencing complex; RNA: RNA-Interferenz (RNA-Silencing); ZFP: Zink-Finger-Protein
Behandlung mit intrathekal applizierten Medikamenten: ASOs
Die Einführung moderner genetischer Methoden hat das Gebiet der neurodegenerativen und neuromuskulären Erkrankungen durch die Entschlüsselung der zugrunde liegenden Pathomechanismen revolutioniert. Dennoch war die Auswahl an effektiven Behandlungsmöglichkeiten bisher begrenzt. Dies änderte sich, als Antisense-Oligonukleotide (ASOs) aus der präklinischen Forschung in die klinische Praxis überführt wurden. Im Jahr 2016 wurden zwei ASOs von der US Food and Drug Administration (FDA) für klinische Studien zugelassen und zeigten eine bemerkenswerte Wirksamkeit bei Patienten mit Duchenne-Muskeldystrophie (DMD) und besonders mit spinaler Muskelatrophie (SMA) [52].
Im Bereich der Huntington-Forschung bieten ASOs einen Ansatz zur Reduktion des krankheitsverursachenden mutierten HTT-Proteins [20, 34], indem sie sequenzspezifisch an die HTT-prä-mRNA im Zellkern binden, woraufhin eine Ribonuklease-H-vermittelte Hydrolyse des RNA-Strangs mit anschließender Verarbeitung der Spaltprodukte erfolgt [18, 50].
Die beiden HTT-Allele können sich aufgrund vereinzelter Polymorphismen in der Nukleotidsequenz (sog. SNPs single nucleotide polymorphisms) unterscheiden. Dies kann therapeutisch bei der Auswahl der ASOs berücksichtigt werden [37]. Nicht-allel-selektive ASOs vermindern sowohl die Expression des physiologischen als auch des mutanten HTT-Proteins [20, 50]. Die vollständige Hemmung der gesamten HTT-Expression ist aufgrund der zahlreichen zellulären Funktionen des HTTs im gesamten Organismus nicht anzustreben [8]. Erste Daten zeigen, dass eine Reduktion der Gen-Expression um die Hälfte möglich zu sein scheint, ohne die Zellfunktion zu stören [32]. Es gibt jedoch weitere Problemstellungen, die sich vor allem auf die Verabreichung der ASOs beziehen. Zum einen muss die Verabreichung intrathekal erfolgen, weil die ASO-basierten Therapeutika die Blut-Hirn-Schranke nicht passieren können. Darüber hinaus ist eine wiederholte Verabreichung im Abstand von zwei bis vier Monaten notwendig, da ASOs über körpereigene Nukleasen abgebaut werden [36] und somit nur zu einer vorübergehenden Reduktion des mHTT-Spiegels führen. Einerseits kann diese nur vorübergehende Wirksamkeit von Vorteil sein, wenn die Therapie aufgrund von Nebenwirkungen abgebrochen oder pausiert werden muss. Andererseits stellt die wiederholte intrathekale Injektion eine Belastung für die Patienten und die Versorgungsstrukturen dar. Darüber hinaus ist die unterschiedliche Verteilung von ASOs über den Liquor cerebrospinalis im Gehirngewebe zu erwarten; dieser Aspekt der Pharmakokinetik ist bisher nur unzureichend geklärt [19]. Erste Hinweise auf eine unzureichende Verteilung von intrathekal injizierten ASOs im Gehirn gab eine präklinische Studie mit Primaten. Diese konnte zwar eine anhaltende Reduktion der HTT-mRNA in den meisten Hirn- und Rückenmarksregionen nachweisen, das Ausmaß der Suppression fiel jedoch in tieferen Hirnstrukturen (z. B. in den Basalganglien) geringer aus als in oberflächlichen Kortex- und Rückenmarksregionen [4, 17].
Nicht-selektive ASOs
Die nicht-allel-selektiven ASOs reduzieren sowohl das mutierte mHTT als auch das physiologische HTT [2]. Die erste klinische Arzneimittelprüfung zum Nachweis der Wirksamkeit (Phase III) intrathekal verabreichter nicht-allel-selektiver ASOs für die HK wurde im Jahr 2019 von der Firma Hoffmann-La Roche gestartet [40]. Diese Phase-III-Studie (Generation-HD1; NCT03761849) folgt auf die weiterhin laufende Phase-I/IIa-Erweiterungsstudie (GenExtend; NCT03842969) und untersucht die Wirksamkeit des nicht-selektiven HTT-ASOs RO7234292 (RG6042) [40]. Zuvor konnte für das Medikament in der Phase-Ib/IIa-Studie gute Verträglichkeit und Sicherheit nachgewiesen werden und darüber hinaus zeigte sich eine dosisabhängige Senkung der mHTT-Konzentration im Liquor um bis zu 42 % [40], sodass eine größere multizentrische Studie mit mehr als 800 Patienten weltweit initiiert wurde. Leider führte eine Datenanalyse des unabhängigen Datenüberwachungskomitees (iDMC) im März 2021 zu der Empfehlung, die Studie in allen Dosen abzubrechen, da das Studienmedikament bei behandelten Patienten im Vergleich zur Placebo-Gruppe zur Progression der erfassten klinisch-funktionellen, motorischen und kognitiven Funktionen führte, also einen negativen Effekte hatte [16a, 30]. Eine detaillierte Auswertung der bisherigen Studiendaten lag zum Zeitpunkt der Einreichung dieser Übersichtsarbeit von Hoffmann-La Roche noch nicht vor. Die Studie ist zurzeit pausiert und weitere Prüfungen der Sicherheit und Wirksamkeit werden vorgenommen, jedoch ohne dass die Studienmedikation weiter verabreicht wird.
Allel-selektive ASOs
Klinische Studien zu weiteren Behandlungsansätzen mit allel-selektiven ASOs sowie zu gentherapeutischen Ansätzen mit RNA-Molekülen werden aktuell in präklinischen und klinischen Phase-I/II-Studien untersucht [35]. Die Firma Wave Life Sciences startete im Jahr 2017 zwei Phase-Ib/IIa-Studien Precision-HD1 (NCT03225833) und -HD2 (NCT03225846), um die Sicherheit und Verträglichkeit ihrer beiden allel-selektiven ASOs (WVE-120 101 und WVE-120 102) zu untersuchen. Diese gezielte allel-selektive Reduktion der mHTT wird durch Bindung des ASO an Einzelnukleotid-Polymorphismen (SNPs) erreicht, die nur auf dem erweiterten mutierten HTT-Gen vorhanden sind [27]. Die SNPs kommen in unterschiedlichen ethnischen Gruppen in verschiedener Frequenz vor, sodass dieser Ansatz zunächst ein Screening der Patienten auf heterozygotes Vorliegen des entsprechenden SNPs erfordert. Nach Angaben von Wave sind SNP 1 und SNP 2, wie sie in Precision-HD1 und -HD2 verwendet werden, bei etwa 50 % bzw. 40 % aller HK-Patienten weltweit vorhanden [27]. Auch diese beiden Studien wurden im März 2021 wegen mangelnder Wirksamkeit des Studienmedikaments im Vergleich zu Placebo vorzeitig abgebrochen. Eine Zwischenauswertung ergab, dass im Liquor keine signifikante und klinisch relevante Senkung der mHTT-Konzentration erreicht werden konnte. Derzeit setzt Wave in einer weiteren Phase-II-Studie (Precision-HD3) ein verändertes allel-selektives ASO (WVE-003) für SNP3 ein, das bei ungefähr 40 % aller HK-Patienten vorhanden ist.
Behandlung mit oralen niedermolekularen Spleißmodulatoren
Eine weitere Behandlungsoption, die ebenfalls über den mRNA-Abbau wirkt, sind oral verfügbare, niedermolekulare Spleißmodulatoren. Diese greifen direkt in die Transkription des HTT-Gens ein, indem sie ein Pseudoexon mit vorzeitigem Stopp-Codon in der prä-mRNA aktivieren, was zum Abbau der HTT-mRNA und so zur Reduktion der HTT-Produktion führt (Abb. 2). Ähnlich wie bei der ASO-basierten Therapie können Spleißmodulatoren lediglich zu einer transienten Reduktion von mHTT führen, weshalb eine wiederholte Anwendung für eine langfristigere Wirkung erforderlich ist [42]. Im Gegensatz zur ASO-basierten Therapie stellt die orale Verfügbarkeit eine wesentlich geringere Belastung und eine weniger invasive Behandlung dar. Aufgrund ihres Wirkungsmechanismus sind Spleißmodulatoren auch für die Behandlung anderer neurodegenerativer Erkrankungen geeignet. Zum Beispiel werden sie im Kontext der spinalen Muskelatrophie (SMA) bereits in präklinischen und klinischen Studien untersucht. Die beiden Firmen Novartis und PTC Therapeutics sind hierbei maßgeblich an der Entwicklung und Untersuchung von Spleißmodulatoren beteiligt. Im Kontext der HK wird Novartis voraussichtlich ab Ende dieses Jahres eine erste klinische Phase-IIb-Studie zur Untersuchung der Sicherheit und Verträglichkeit von Branaplam (HTT-001) bei erwachsenen HK-Patienten durchführen. Obwohl Branaplam eigentlich zur Behandlung der SMA entwickelt wurde, hatten Daten aus einer aktuellen Phase-I-Studie (NCT02268552) mit SMA-Patienten gezeigt, dass Branaplam zu einer Reduktion von HTT-mRNA führte [24].
Behandlung mit RNA-Interferenz-basierten Strategien
RNA-Interferenz(RNAi)-basierte Behandlungsstrategien wirken über kurze Nukleotid-Sequenzen, die an komplementäre HTT-mRNA-Sequenzen binden und diese anschließend über die RNase im RNA-induced Silencing Complex (RISC) abbauen (Abb. 2) [50]. Es gibt verschiedene RNAi-Moleküle, darunter small interfering RNA (siRNA), short hairpin RNA (shRNA) und microRNA (miRNA). Da sich shRNA und miRNA unzureichend im Hirngewebe verteilen, müssen sie mittels Adeno-assoziierten Virus(AAV)-Vektoren [39, 50] oder mittels lentiviraler Vektoren [3] stereotaktisch ins Hirnparenchym injiziert werden. RNAi-basierte Strategien können sowohl nicht-spezifisch die HTT-mRNA abbauen als auch allel-spezifisch [27] gezielt nur die mHTT-RNA abbauen. Mit Blick auf klinische Studien haben zwei Firmen, uniQure und Voyager, bereits die Zulassung zur klinischen Erprobung ihrer RNAi-basierten Gentherapien von der FDA erhalten. Beide Ansätze wirken nicht-allel-spezifisch, verwenden miRNA als RNAi-Molekül und einen rekombinanten AAV-Vektor für die intraparenchymatöse Verabreichung.
Die rekombinante AAV-basierte Gentherapie (rAAV5-miHTT; AMT-130) von uniQure exprimiert eine miRNA, die speziell dafür entwickelt wurde, an das HTT-Exon 1 zu binden [23]. In präklinischen Studien in In-vitro- [16] und HK-Tiermodellen konnte die Sicherheit und Wirksamkeit von AMT-130 bereits nachgewiesen werden [9, 38]. Hierbei zeigten sich eine gute, dosisabhängige Verteilung im gesamten Hirngewebe sowie eine statistisch bedeutsame mHTT-Reduktion in allen injizierten Regionen [9]. Diese vielversprechenden Ergebnisse konnten in einer weiteren Studie mit transgenen HK-Minipigs bestätigt werden: Es zeigte sich nach Injektion von AMT-130 in den Nucleus Caudatus und das Putamen eine gute Verteilung in diesen Zielregionen sowie Ausbreitung in angrenzende Gehirnareale (Amygdala, Kortex und Thalamus) mit einer daraus resultierenden Reduktion des mHTT-Proteins, die auch noch nach mehreren Monaten im Liquor messbar war [46]. Seit dem letzten Jahr wird AMT-130 bereits im Rahmen einer Phase-I/II-Studie an amerikanischen HK-Patienten untersucht (HD.GeneTRX1, NCT04120493). Darüber hinaus wird demnächst eine Open-Label-Extension-Studie (HD.GeneTRX2) mit europäischen HK-Patienten mit AMT-130 starten.
Präklinische Therapieansätze zur Modulation der HTT-Transkription oder HTT-Gen-Editierung
Darüber hinaus befinden sich zwei innovative Therapieansätze in der präklinischen Phase. Beide Ansätze greifen im Wesentlichen bereits auf der DNA-Ebene ein – jedoch mit unterschiedlichem molekularem Mechanismus. Die Zink-Finger-Protein-Repressor-Komplexe (ZFP) verringern selektiv die Transkription des mutanten HTT-Allels, indem sie direkt an die verlängerte CAG-Sequenz binden und so die Transkription hemmen [54].
CRISPR/Cas9 kann bei entsprechender Konstruktion direkt an der CAG-Expansionsmutation ansetzen, um diese mittels Genom-Editierung zu modifizieren. Eine Leit-RNA-Sequenz bindet das Cas9 an die entsprechende Zielsequenz (hier: mutantes HTT) und führt dort zum Doppelstrangbruch. Daraufhin kann die entsprechende DNA-Sequenz editiert oder vollständig inaktiviert werden [33].
Fazit
In den letzten Jahren wurden mehrere klinische Studien zur Sicherheit, Verträglichkeit und Wirksamkeit neuer therapeutischer Ansätze durchgeführt. Diese Ansätze eröffnen die Möglichkeit, die klinische Manifestation der Huntington-Krankheit zu verzögern oder das Auftreten von Symptomen zu verhindern, indem frühzeitig in prämanifesten Stadien der Erkrankung eingegriffen wird. Da bisherige Studien die erhofften Ergebnisse nicht erzielen konnten, bedarf es einer weiteren Entwicklung der hier genannten Therapien sowie eines besseren Verständnisses der komplexen physiologischen Funktion des Huntingtin-Proteins. Unabhängig davon stellt die dynamische Entwicklung innovativer Behandlungsansätze die bestehende Versorgungsstruktur mit spezialisierten HK-Zentren vor große Herausforderungen. Sie erfordert die Implementierung geeigneter neuer Versorgungspfade und kooperativer Versorgungsmodelle, um den Anforderungen, welche die Bereitstellung krankheitsmodifizierender Therapien für Betroffene der HK mit sich bringt, gerecht zu werden.
Interessenkonflikterklärung
KSL, WF, AM, RH, ACL geben keine Interessenkonflikte an.
Literatur
1. Bates GP, Dorsey R, Gusella JF, Hayden MR, et al. Huntington disease. Nat Rev Dis Primers 2015;1:15005.
2. Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med 2019;70:307–21.
3. Cambon K, Zimmer V, Martineau S, Gaillard M-C, et al. Preclinical evaluation of a lentiviral vector for Huntingtin silencing. Mol Ther Methods Clin Dev 2017;5:259–76.
4. Caron NS, Dorsey ER, Hayden MR. Therapeutic approaches to Huntington disease: from the bench to the clinic. Nat Rev Drug Discov 2018;17:729–50.
5. Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci 2005;6:919–30.
6. Chaganti SS, McCusker EA, Loy CT. What do we know about Late Onset Huntington’s Disease? J Huntington’s Dis 2017;6:95–103.
7. DiFiglia M, Sapp E, Chase KO, Davies SW, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997;277:1990–3.
8. Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet 2000;26:300–6.
9. Evers MM, Miniarikova J, Juhas S, Vallès A, et al. AAV5-miHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington’s disease minipig model. Mol Ther 2018;26:2163–77.
10. Gardiner SL, Boogaard MW, Trompet S, De Mutsert R, et al. Prevalence of carriers of intermediate and pathological polyglutamine disease-associated alleles among large population-based cohorts. JAMA Neurology 2019;76:650.
11. Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium, Lee J-M, Correia K, Loupe J, et al. Huntington’s disease onset is determined by length of uninterrupted CAG, not encoded polyglutamine, and is modified by DNA maintenance mechanisms. bioRxiv 2019:529768.
12. Gipson TA, Neueder A, Wexler NS, Bates GP, et al. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol 2013;10:1647–52.
13. Harding RJ, Loppnau P, Ackloo S, Lemak A, et al. Design and characterization of mutant and wildtype huntingtin proteins produced from a toolkit of scalable eukaryotic expression systems. J Biol Chem 2019;294:6986–7001.
14. Huntington’s disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993;72:971–83.
15. Kennedy L, Shelbourne PF. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington’s disease? Hum Mol Genet 2000;9:2539–44.
16. Keskin S, Brouwers CC, Sogorb-Gonzalez M, Martier R, et al. AAV5-miHTT lowers Huntingtin mRNA and protein without off-target effects in patient-derived neuronal cultures and astrocytes. Mol Ther Methods Clin Dev 2019;15:275–84.
16a. Kingwell K. Double setback for ASO trials in Huntington disease. Nat Rev Drug Discov 2021;20:412–3.
17. Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012;74:1031–44.
18. Larrouy B, Blonski C, Boiziau C, Stuer M, et al. RNase H-mediated inhibition of translation by antisense oligodeoxyribonucleotides: use of backbone modification to improve specificity. Gene 1992;121:189–94.
19. Leavitt BR, Kordasiewicz HB, Schobel SA. Huntingtin-lowering therapies for Huntington disease: a review of the evidence of potential benefits and risks. JAMA Neurol 2020;77:764–72.
20. Leavitt BR, Tabrizi SJ. Antisense oligonucleotides for neurodegeneration. Science 2020;367:1428–9.
21. Lee J, Correia K, Loupe J, Kim K-H, et al. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 2019;178:887–900.e14.
22. Lee JM, Ramos EM, Lee JH, Gillis T, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 2012;78:690–5.
23. Miniarikova J, Zanella I, Huseinovic A, van der Zon T, et al. Design, characterization, and lead selection of therapeutic miRNAs targeting Huntingtin for development of gene therapy for Huntington’s disease. Mol Ther Nucleic Acids 2016;5:e297.
24. Novartis. Novartis receives US Food and Drug Administration (FDA) Orphan Drug Designation for branaplam (LMI070) in Huntington’s disease (HD). Basel: Novartis, 2020. https://www.novartis.com/news/media-releases/novartis-receives-us-food-and-drug-administration-fda-orphan-drug-designation-branaplam-lmi070-huntington%27s-disease-hd (Zugriff am 31.07.2021).
25. Paulsen J, Miller A, Hayes T, Shaw E. Cognitive and behavioral changes in Huntington disease before diagnosis. Handb Clin Neurol 2017;144:69–91.
26. Paulsen JS, Langbehn DR, Stout JC, Aylward E, et al. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008;79:874–80.
27. Pfister EL, Kennington L, Straubhaar J, Wagh S, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol 2009;19:774–8.
28. Quarrell OWJ, Nance MA, Nopoulos P, Reilmann R, et al. Defining pediatric Huntington disease: time to abandon the term juvenile Huntington disease? Mov Disord 2019;34:584–5.
29. Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, et al. The prevalence of Huntington’s disease. Neuroepidemiology 2016;46:144–53.
30. Roche. Roche provides update on tominersen programme in manifest Huntington’s disease. Basel: Hoffmann-La-Roche, 2021. https://www.roche.com/dam/jcr:e077be26-41a0-4431-ae19-8f8dc846179a/en/22032021-mr-update-on-tominersen-programme-en.pdf (Zugriff am 31.07.2021).
31. Rodrigues FB, Wild EJ. Huntington’s disease clinical trials corner: February 2018. J Huntingtons Dis 2018;7:89–98.
32. Saudou F, Humbert S. The biology of Huntingtin. Neuron 2016;89:910–26.
33. Shin JW, Kim KH, Chao MJ, Atwal RS, et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 2016;25:4566–76.
34. Silva AC, Lobo DD, Martins IM, Lopes SM, et al. Antisense oligonucleotide therapeutics in neurodegenerative diseases: the case of polyglutamine disorders. Brain 2020;143:407–29.
35. Skotte NH, Southwell AL, Østergaard ME, Carroll JB, et al. Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS ONE 2014;9:e107434.
36. Smith CIE, Zain R. Therapeutic oligonucleotides: state of the art. Annu Rev Pharmacol Toxicol 2019;59:605–30.
37. Southwell AL, Skotte NH, Kordasiewicz HB, Østergaard ME, et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther 2014;22:2093–106.
38. Spronck EA, Vallès A, Lampen MH, Montenegro-Miranda PS, et al. Intrastriatal administration of AAV5-miHTT in non-human primates and rats is well tolerated and results in miHTT transgene expression in key areas of Huntington disease pathology. Brain Sci 2021;11:129.
39. Stanek LM, Sardi SP, Mastis B, Richards AR, et al. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther 2014;25:461–74.
40. Tabrizi SJ, et al. Inhibition of mutant huntingtin expression in a randomized trial of patients with Huntington’s disease. N Engl J Med 2019;380:2307–16.
41. Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol 2020;16:529–46.
42. Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 2019;101:801–19.
43. Tabrizi SJ, Scahill RI, Durr A, Roos RAC, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011;10:31–42.
44. Tabrizi SJ, Scahill RI, Owen G, Durr A, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 2013;12:637–49.
45. Travessa AM, Rodrigues FB, Mestre TA, Ferreira JJ. Fifteen years of clinical trials in Huntington’s disease: a very low clinical drug development success rate. J Huntingtons Dis 2017;6:157–63.
46. Vallès A, Evers MM, Stam A, Sogorb-Gonzalez M, et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci Transl Med 2021;13;eabb8920; doi: 10.1126/scitranslmed.abb8920.
47. Walker FO. Huntington’s disease. Lancet 2007;369:218–28.
48. Wang N, Gray M, Lu X-H, Cantle JP, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med 2014;20:536–41.
49. Wanker EE, Ast A, Schindler F, Trepte P, et al. The pathobiology of perturbed mutant huntingtin protein – protein interactions in Huntington’s disease. J Neurochem 2019;151:507–19.
50. Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol 2017;16:837–47.
51. Wright GEB, Collins JA, Kay C, McDonald C, et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet 2019;104:1116–26.
52. Wurster CD, Ludolph AC. Antisense oligonucleotides in neurological disorders. Ther Adv Neurol Disord 2018;11:175628641877693.
53. Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000;101:57–66.
54. Zeitler B, Froelich S, Marlen K, Shivak DA, et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat Med 2019;25:1131–42.
Prof. Dr. Dr. med. Albert C. Ludolph, Universitätsklinikum Ulm, Abteilung Neurologie, Oberer Eselsberg 45, 89081 Ulm, Deutsches Zentrum für Neurodegenerative Erkrankungen, Ulm, E-Mail: albert.ludolph@uni-ulm.de
Wiebke Frank, M. Sc., Dr. rer. nat. Katrin S. Lindenberg, Universitätsklinikum Ulm, Abteilung Neurologie, Oberer Eselsberg 45, 89081 Ulm
Med. Univ. Dr. Alzbeta Mühlbäck, Universitätsklinikum Ulm, Abteilung Neurologie, Oberer Eselsberg 45, 89081 Ulm; Huntington-Zentrum Süd, kbo-Isar-Amper-Klinikum, Taufkirchen (Vils); Klinik für Neurologie und Zentrum für klinische Neurowissenschaften, 1. Medizinische Fakultät, Karlsuniversität, Prag
Dr. med. Rainer Hoffmann, Huntington-Zentrum Süd, kbo-Isar-Amper-Klinikum, Taufkirchen (Vils)
Gene therapy approaches in Huntington’s disease – Review and outlook on current gene-modifying approaches
Huntington’s disease (HD) is an autosomal dominant hereditary neurodegenerative disorder characterised by progressive impairments in motor function, behaviour and cognition, leading to disability, dependency and premature death. Due to its monogenetic origin, HK serves as a model disease for many rare diseases and is currently the focus of general medical interest as numerous clinical trials have been conducted. Insights into the pathogenesis of HD in the 28 years since the identification of the huntingtin (HTT) gene and the HD mutation have enabled the identification of a number of different molecular targets and the development of novel, innovative therapeutic strategies that target different potential points of intervention in the pathogenetic chain – starting with the mutated HTT gene. The authors provide an overview of novel treatment strategies from current clinical trials and from previously completed studies. These strategies aim to decrease the production of the mutant HTT gene products by transcriptional repression or to decrease the production of mHTT by modulating proteostasis, e.g., autophagy or the ubiquitin-proteasome system. Transcriptional repression can be realised by reducing the levels of different mRNA species encoding HTT (pre-mRNA or mature mRNA), using either DNA-based compounds (antisense oligonucleotides – ASOs), RNA-based therapeutics (miRNA, siRNA), or small molecules acting via translational RNA decay to induce transcriptional repression either allele-selectively or non-allele-selectively. In addition, strategies aimed at correcting CAG expansion mutation at the DNA level by gene editing (e.g., using CRISPR/Cas9) or preventing somatic expansions or inducing somatic contractions are currently under investigation.
Key words: Huntingtin, antisense oligonucleotides, zinc-finger repressor complexes, genome editing, CRISPR/Cas9, gene therapy, adeno-associated viruses, small molecule splicing modulators
Psychopharmakotherapie 2021; 28(05):187-193