Holger Petri, Bad Wildungen*
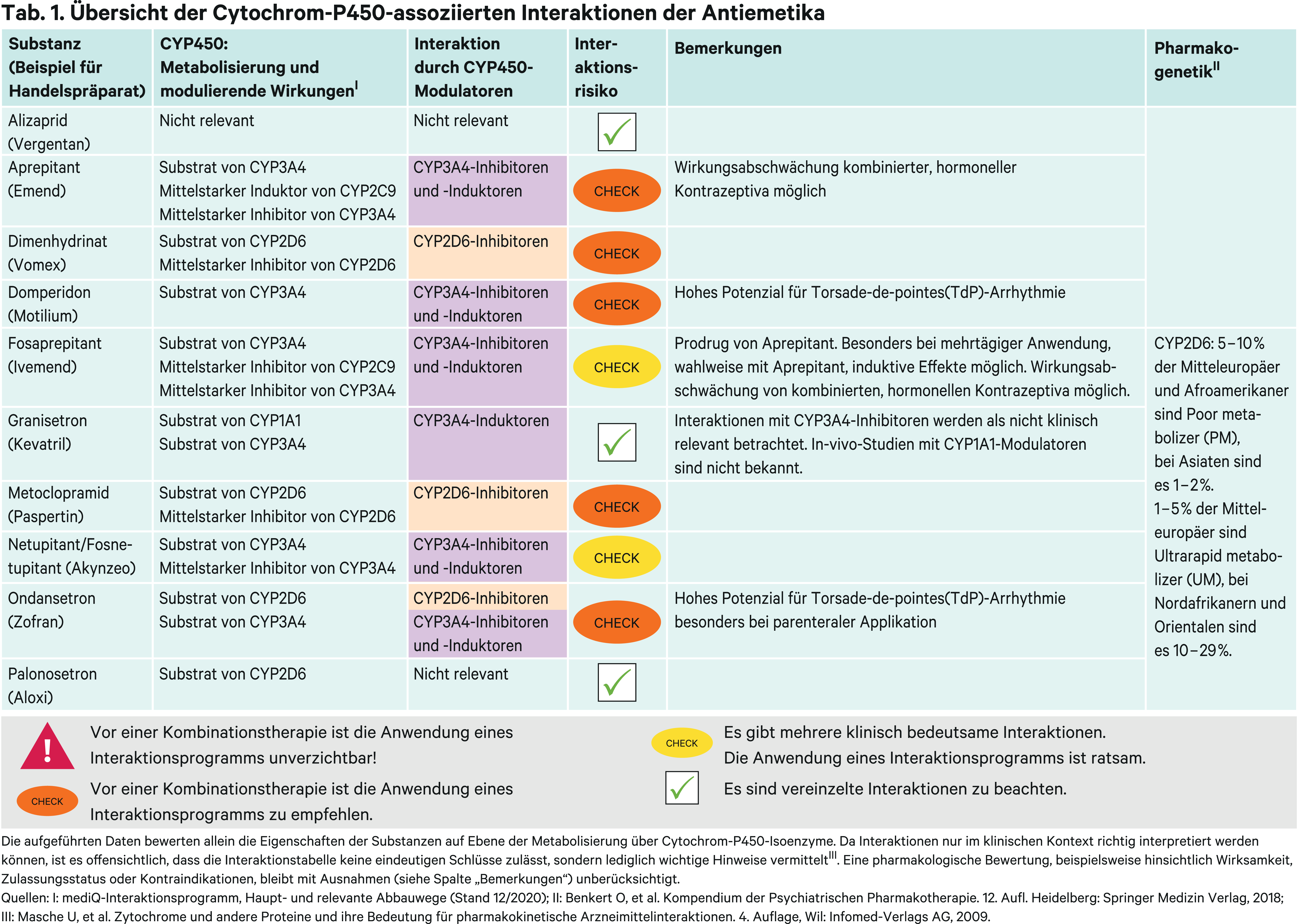
5-HT3-Rezeptorantagonisten (Setrone)
Ondansetron
Der Serotonin(5-HT)-3-Rezeptorantagonist Ondansetron wird über das polymorph exprimierte Cytochrom-P450(CYP)-Isoenzym 2D6 sowie CYP1A2 und 3A4 zu inaktiven Metaboliten abgebaut [2].
Patienten mit einem Poor-Metabolizer(PM)-Phänotyp benötigen wegen der unveränderten Eliminationshalbwertszeit von Ondansetron gegenüber normalen Verstoffwechslern keine Änderung der Dosis oder Dosisfrequenz [19]. Der Abbau wird in dieser Situation durch CYP1A2 und 3A4 kompensiert [2]. Somit sollten CYP2D6-Inhibitoren (Abb. 1) auch keine Auswirkungen auf die Exposition von Ondansetron haben. Ultraschnelle Metabolisierer (UM) von CYP2D6-Substraten zeigen eine erhöhte Stoffwechselaktivität. In einer Studie wurden 250 Patientinnen, die einer Allgemeinanästhesie unterzogen worden, genotypisiert. Sie erhielten zur Prophylaxe von postoperativer Übelkeit und Erbrechen (PONV) eine Dosis von 4 mg Ondansetron. Der Anteil der Frauen mit Erbrechen lag bei Trägerinnen des CYP2D6-UM-Phänotyps mit 45 % dreimal so hoch im Vergleich zu denen mit normalem CYP2D6-Phänotyp (Extensive Metabolizer) [5]. Solche Patienten sollten alternativ ein Setron wie Granisetron erhalten, da dessen Biotransformation CYP2D6-unabhängig erfolgt [2]. Die Bildung von CYP2D6-Enzymen ist durch Xenobiotika nicht induzierbar [34].
In einer Untersuchung mit gesunden Probanden senkte der CYP3A4-Induktor Rifampicin die AUC (Fläche unter der Konzentrations-Zeit-Kurve) von Ondansetron bei peroraler Gabe um 65 % und bei parenteraler Applikation um etwa die Hälfte [30]. Die Komedikation mit Rifampicin und anderen starken CYP3A4-Induktoren (Abb. 1) kann zu einer verminderten antiemetischen Wirksamkeit von Ondansetron führen [30]. Ondansetron verlängert dosisabhängig das frequenzkorrigierte QT-Intervall (QTc) im Elektrokardiogramm, was zu potenziell lebensbedrohlichen Torsade-de-pointes(TdP)-Arrhythmien führen kann. Intravenös soll daher Ondansetron initial höchstens mit 16 mg angewendet werden, bei Patienten ab 75 Jahren mit 8 mg [19]. Deshalb ist Vorsicht geboten, wenn potente CYP3A4-Hemmer kombiniert werden. In der Regel ist der CYP2D6-Phänotyp des Patienten unbekannt. Bei verminderter CYP2D6-Stoffwechselkapazität (Intermediate und Poor Metabolizer) kann es zu einem relevanten Plasmaspiegelanstieg kommen, da zwei Abbauwege blockiert sind.
Granisetron
Granisetron wird über CYP3A4 demethyliert und über CYP1A1 zu 7-Hydroxygranisetron abgebaut [2]. Die Metaboliten tragen nur unwesentlich zur antiemetischen Wirkung bei. Die Interaktion mit dem starken CYP3A4-Hemmer Ketoconazol wird als vermutlich klinisch nicht relevant gewertet [14]. Studien zu Interaktionen mit CYP3A4-Induktoren sind nicht bekannt. Eine Wirkungsabschwächung kann jedoch nicht ausgeschlossen werden und bei Therapieversagen sollte eine Dosissteigerung oder ein Wechsel auf Palonosetron erwogen werden, dessen Exposition durch Rifampicin vernachlässigbar beeinflusst wird [4]. Omeprazol kann in vitro die Bildung von CYP1A1 induzieren [32]. In-vivo-Daten zu einer möglichen Interaktion von Omeprazol und anderen Modulatoren mit CYP1A1-Substraten sind bisher in Studien nicht dokumentiert [23].
Palonosetron
Palonosetron ist primär ein Substrat von CYP2D6. Der metabolische Abbau über andere Enzyme ist nachgeordnet [2, 11]. Die Hälfte einer Dosis unterliegt der Biotransformation, wobei die Metaboliten nicht zur klinischen Wirksamkeit beitragen. Etwa 40 % der Dosis werden renal unverändert ausgeschieden [11]. In einer pharmakokinetischen Analyse wurde gezeigt, dass starke CYP2D6-Inhibitoren wie Fluoxetin und Paroxetin (Abb. 1) keine signifikante Auswirkung auf die Clearance von Palonosetron haben [11]. Wie sich ein UM-Phänotyp auf das therapeutische Ansprechen von Palonosetron auswirkt, ist nicht bekannt.
Neurokinin-1-Rezeptor-Antagonisten
Aprepitant
Aprepitant zeigt ein komplexes pharmakokinetisches Interaktionspotenzial. Es ist sowohl ein Substrat („Victim Drug“) als auch ein „Perpetrator“ der Enzyme CYP2C9 und 3A4. Durch Ketoconazol stieg die AUC einer 125-mg-Einzeldosis Aprepitant um das 5-Fache. Rifampicin reduzierte die Exposition einer 375-mg-Einzeldosis Aprepitant um 91 %. Starke CYP3A4-Induktoren sollten in Kombination mit Aprepitant gemieden werden und starke Inhibitoren sind nur mit Vorsicht anzuwenden [12].
Aprepitant ist ein moderater CYP3A4-Inhibitor. Die AUC-Werte von oral eingenommenem Dexamethason und Methylprednisolon stiegen bei Probanden um das 2,2- bzw. 2,5-Fache [1]. Folglich sollten die oralen Dosen von Dexamethason bei einem 3-Tages-Therapieschema mit Aprepitant um etwa die Hälfte reduziert werden [12]. Bei anderen CYP3A4-Substraten wie Bosutinib und Midazolam erhöhte sich die Exposition um das 2,0- bzw. 3,3-Fache, beim hochpotenten Opioid Oxycodon um 25 % [1, 20]. In einem Fallbericht reagiert ein Patient unter niedrig dosiertem Quetiapin nach einer Aprepitant-Antiemese mit tiefer Somnolenz. Die gemessenen Talspiegel waren 11-fach erhöht [29].
Auch bei einem 3-Tages-Therapieschema kommt es zu einer Induktion von CYP-Enzymen. So senkte Aprepitant die AUC des CYP2C9-Testsubstrats Tolbutamid an Tag 4 um 23 % und an Tag 8 um 27 % [1].
Warfarin besteht als racemisches Gemisch zu gleichen Teilen aus S- und R-Warfarin. S-Warfarin besitzt eine 2- bis 5-fach höhere blutgerinnungshemmende Aktivität als R-Warfarin. S-Warfarin wird primär über CYP2C9 abgebaut [26]. In einer Studie mit gesunden Freiwilligen sanken die Talspiegel von S-Warfarin an den Tagen 5 bis 8 um bis zu 34 %. An Tag 8 war der INR-Wert um 11 % niedriger als in der Kontrollgruppe [1]. Entsprechende Vorsicht ist geboten bei Substraten von CYP2C9 mit enger therapeutischer Breite wie Phenytoin [12].
Die CYP3A4-Induktion kann im Einzelfall von klinischer Relevanz sein. Aprepitant reduzierte die AUC von oralem Midazolam als CYP3A4-Testsubstrat in pharmakokinetischen Studien um lediglich maximal 19 % an Tag 8.
Jedoch sank die AUC des Estrogens Ethinylestradiol, einem CYP3A4-Substrat, in einer klinischen Studie um 43 % [1]. Die Minimalkonzentrationen von Ethinylestradiol waren um 64 % niedriger im Vergleich zur Kontrollgruppe, die des Gestagens Norethisteron um 60 %. Die Wirksamkeit hormoneller Kontrazeptiva kann somit während und nach der Einnahme von Aprepitant vermindert sein. Alternative, nichthormonelle unterstützende Maßnahmen sollten während der Behandlung mit Aprepitant und während der letzten zwei Monate nach der letzten Dosis ergriffen werden [12].
Fosaprepitant
Fosaprepitant ist ein Prodrug von Aprepitant und damit sind dieselben Interaktionen mit CYP3A4-Modulatoren zu erwarten wie mit Aprepitant [13]. Womöglich sind diese bei parenteraler Gabe wegen des fehlenden First-Pass-Effekts geringer ausgeprägt. Fosaprepitant hat auch CYP3A4-hemmende Eigenschaften und erhöhte die AUC von Dexamethason um das Doppelte und von Midazolam um das 1,8-Fache [13].
Wegen der nur einmaligen Anwendung bei Erwachsenen sind induktive Effekte auf CYP-Enzyme nicht zu erwarten. So war die AUC von oralem Midazolam an Tag 4 unverändert. Dennoch sollten, wie bei Aprepitant, alternative nichthormonelle unterstützende Maßnahmen zur Verhütung ergriffen werden [13]. Dies gilt umso mehr, wenn sich bei Jugendlichen die antiemetische Therapie über drei Tage, wahlweise an Tag 2 und 3 auch mit Aprepitant, erstrecken kann und dann induktive Effekte möglich sind.
Netupitant/Fosnetupitant
Netupitant und sein parenterales Prodrug Fosnetupitant sind nur in Fixkombination mit Palonosetron verfügbar [10]. Der metabolische Abbau wird primär über CYP3A4 vermittelt. Es entstehen drei aktive Metaboliten mit einer geringeren Exposition verglichen mit der Muttersubstanz [4]. Ketoconazol erhöhte die AUC von Netupitant in einer pharmakokinetischen Studie mit gesunden Probanden um das 2,4-Fache, Rifampicin senkte den AUC-Wert um 83 %. Es kam durch Kombination mit Ketoconazol und Rifampicin zu veränderten AUC-Werten der drei Metaboliten, die klinisch aber von geringer Relevanz sein sollten [4].
So auch durch Netupitant, das ein moderater CYP3A4-Hemmer ist, sind Plasmaspiegeländerungen von CYP3A4-Substraten denkbar. Die AUC-Werte von Dexamethason und Midazolam erhöhten sich um das 2,4-Fache, sodass in den Therapieregimen orales Dexamethason um die Hälfte reduziert angewendet werden sollte [10]. Netupitant induziert keine CYP-Enzyme [10]. Im Gegensatz zu Aprepitant sind keine Wechselwirkungen mit hormonellen Kontrazeptiva beobachtet worden [25].
Beide Neurokinin-1-Rezeptorantagonisten sollten im allgemeinen mit Vorsicht angewendet werden in Kombination besonders mit oral einzunehmenden Arzneimitteln, die eine enge therapeutische Breite haben und primär über CYP3A4 metabolisiert werden [10, 12, 13].
Prokinetika
Metoclopramid
Metoclopramid ist ein Substrat von CYP2D6 [7, 16, 24]. In einer Studie erhöhte der starke CYP2D6-Hemmer Fluoxetin die Exposition von Metoclopramid um 89 % [30]. In einer Kasuistik wird über eine 54-jährige Patientin berichtet, die wegen Magenbeschwerden in Abständen Metoclopramid bei guter Verträglichkeit verordnet bekommen hatte. Nach Diagnose einer Major Depression wurde das Antidepressivum Paroxetin angesetzt. Nach Wiederauftreten der Magen-Darm-Symptome bekam sie zweimal die Woche Metoclopramid gespritzt. Sie erlitt hierunter akute extrapyramidal-motorische Nebenwirkungen, die nach intramuskulärer Injektion von Biperiden sistierten. Nach einer Woche verschwanden die extrapyramidal-motorischen Störungen gänzlich. Es wurden zudem stark erhöhte Prolactinspiegel gemessen, die erst nach mehreren Wochen wieder sanken. Die Autoren machen die Kombination aus einem SSRI-Antidepressivum mit Metoclopramid und erhöhte Metoclopramid-Plasmaspiegel verantwortlich für diese Nebenwirkung [22]. Bei schwangeren Frauen mit einem Poor-Metabolizer (PM)-Phänotyp kann das Risiko für eine durch Metoclopramid induzierte motorische Dyskinesie wegen des verlangsamten metabolischen Abbaus erhöht sein [6]. Deshalb sollten potente CYP2D6-Inhibitoren in der Schwangerschaft nur mit Vorsicht mit Metoclopramid kombiniert werden.
In-vitro-Daten zeigen einen inhibitorischen Effekt von Metoclopramid auf die CYP2D6-Aktivität [7, 24]. In-vivo-Daten zum Ausmaß dieser Hemmung sind jedoch nicht bekannt.
Domperidon
Domperidon wird über CYP3A4 metabolisiert. Die starken CYP3A4-Inhibitoren Erythromycin, Itraconazol und Ketoconazol erhöhten die Exposition von Domperidon bei gesunden Probanden um das Dreifache [3, 15, 33]. Folglich sind starke CYP3A4-Hemmer kontraindiziert, da Domperidon dosisabhängig die QT-Zeit verlängern kann und eine TdP-Arrhythmie droht [15]. Der Induktor Rifampicin senkte die Exposition einer Einzeldosis Domperidon um 38 % [9].
Alizaprid
Alizaprid wird nur in geringem Umfang metabolisiert und die Ausscheidung erfolgt hauptpsächlich über die Nieren [17].
Antihistaminika
Dimenhydrinat
Als Antiemetikum findet hauptsächlich Dimenhydrinat Verwendung. Dimenhydrinat dissoziiert im Blut zu Diphenhydramin und 8-Chlortheophyllin [18]. Das Antihistaminikum Diphenhydramin wird gemäß In-vitro-Daten über CYP2D6 verstoffwechselt [27]. In-vivo-Daten hierzu sind nicht bekannt. Schnelle Metabolisierer von CYP2D6 zeigten nach Einnahme von Diphenhydramin paradoxerweise Unruhezustände statt der gewünschten sedierenden Wirkungen. Es wird angenommen, dass die vermehrte Bildung eines aktivierenden Metaboliten Ursache dieser Nebenwirkung ist [8].
In Abhängigkeit vom CYP2D6-Genotyp hemmt Diphenhydramin den Metabolismus von Metoprolol, einem CYP2D6-Substrat, bei männlichen Probanden mit normaler Stoffwechselaktivität um 61 %, bei Personen mit reduzierter Stoffwechselaktivität nur um 10 % [21]. Bei Frauen kann der Effekt ausgeprägter sein. In einer Studie hatten Frauen unter Diphenhydramin einen Anstieg des S-Enantiomers von Metoprolol um 84 %, Männer um 45 %, jeweils mit normalen CYP2D6-Phänotyp [28].
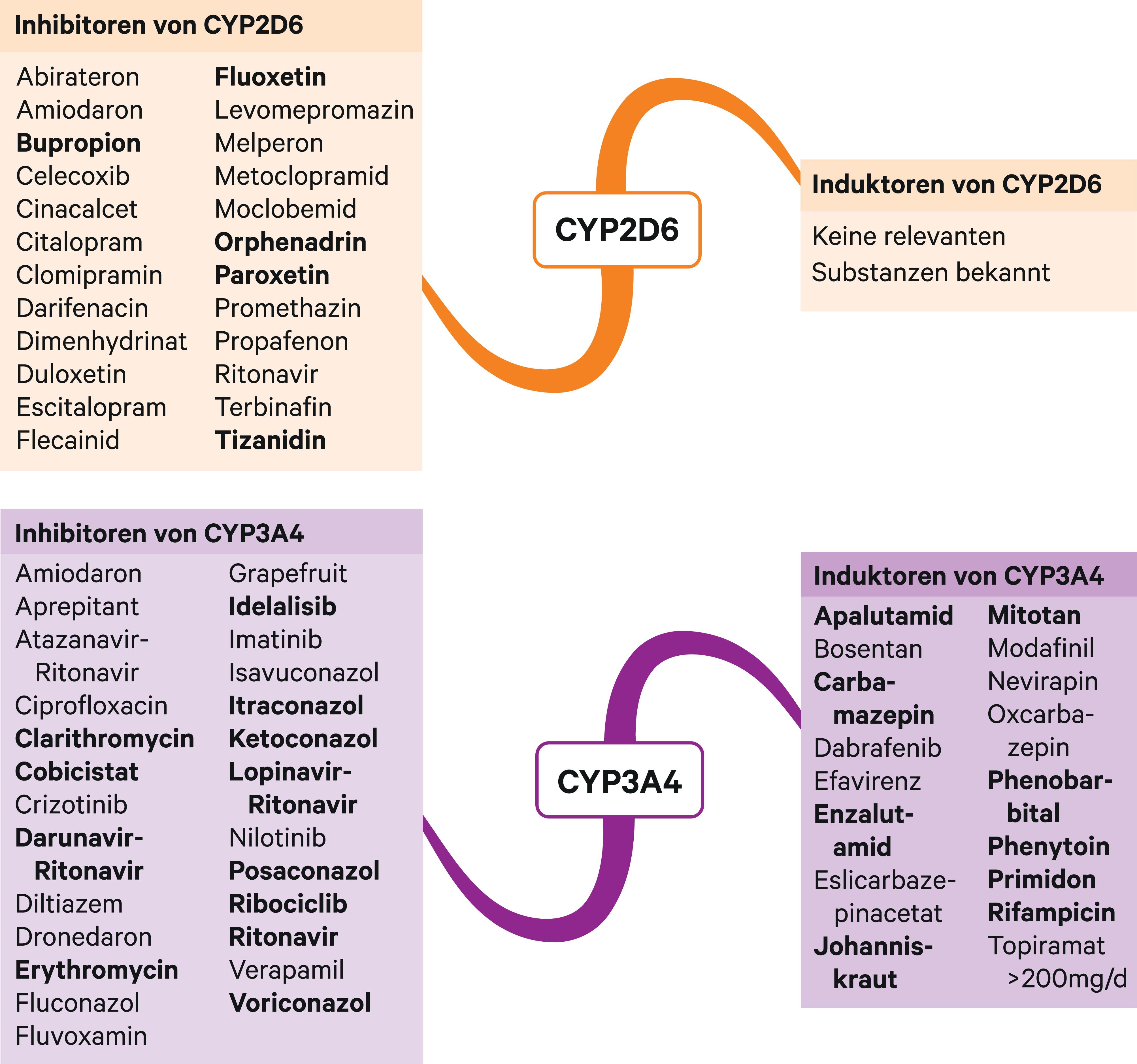
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2D6 und 3A4 (Stand 12/2020) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Aapro MS, Walko CM. Aprepitant: drug-drug interactions in perspective. Ann Oncol 2010;21:2316–23.
2. Bell GC, Caudle KE, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 genotype and use of ondansetron and tropisetron. Clin Pharmacol Ther 2017;102:213–18.
3. Boyce MJ, Baisley KJ, Warrington SJ. Pharmacokinetic interaction between domperidone and ketoconazole leads to QT prolongation in healthy volunteers: a randomized, placebo-controlled, double-blind, crossover study. Br J Clin Pharmacol 2012;73:411–21.
4. Calcagnile S, Lanzarotti C, Rossi G, et al. Effect of netupitant, a highly selective NK₁ receptor antagonist, on the pharmacokinetics of palonosetron and impact of the fixed dose combination of netupitant and palonosetron when coadministered with ketoconazole, rifampicin, and oral contraceptives. Support Care Cancer 2013;21:2879–87.
5. Candiotti KA, Birnbach DJ, Lubarsky DA, et al. The impact of pharmacogenomics on postoperative nausea and vomiting: do CYP2D6 allele copy number and polymorphisms affect the success or failure of ondansetron prophylaxis? Anesthesiology 2005;102:543–9.
6. Chua EW, Harger SP, Kennedy MA. Metoclopramide-Induced Acute Dystonic Reactions May Be Associated With the CYP2D6 Poor Metabolizer Status and Pregnancy-Related Hormonal Changes. Front Pharmacol 2019;10:931.
7. Desta Z, Wu GM, Morocho AM, et al. The gastroprokinetic and antiemetic drug metoclopramide is a substrate and inhibitor of cytochrome P4502D6. Drug Metab Dispos 2002;30:336–43.
8. de Leon J, Nikoloff DM. Paradoxical excitation on diphenhydramine may be associated with being a CYP2D6 ultrarapid metabolizer: three case reports. CNS Spectr 2008;13:133–5.
9. Devandla A, Yamsani SK, Yamsani MR. Effect of rifampicin pretreatment on the oral bioavailability of domperidone in healthy human volunteers. Drug Metab Pers Ther 2015;30:257–61.
10. Fachinformation Akynzeo® Inf./Kps. Stand: Mai/Januar 2020.
11. Fachinformation Aloxi®. Stand: April 2018.
12. Fachinformation Emend®. Stand: August 2020.
13. Fachinformation Ivemend®. Stand: Mai 2020.
14. Fachinformation Kevatril®. Stand: Juni 2020.
15. Fachinformation Motilium®. Stand: Mai 2020.
16. Fachinformation Paspertin®. Stand: November 2019
17. Fachinformation Vergentan®. Stand: April 2019.
18. Fachinformation Vomex®. Stand: Februar 2019
19. Fachinformation Zofran®. Stand: August 2019.
20. Fujiwara Y, Toyoda M, Chayahara N, et al. Effects of aprepitant on the pharmacokinetics of controlled-release oral oxycodone in cancer patients. PLoS One 2014;9:e104215.
21. Hamelin BA, Bouayad A, Méthot J, et al. Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther 2000;67:466–77.
22. Igata R, Hori H, Atake K, et al. Adding metoclopramide to paroxetine induced extrapyramidal symptoms and hyperprolactinemia in a depressed woman: a case report. Neuropsychiatr Dis Treat 2016;12:2279–81.
23. Klomp F, Wenzel C, Drozdzik M, et. al. Drug-drug interactions involving intestinal and hepatic CYP1A enzymes. Pharmaceutics 2020;12(12):1201.
24. Livezey MR, Briggs ED, Bolles AK, et al. Metoclopramide is metabolized by CYP2D6 and is a reversible inhibitor, but not inactivator, of CYP2D6. Xenobiotica 2014;44:309–19.
25. Natale JJ, Spinelli T, Calcagnile S, et al. Drug-drug interaction profile of components of a fixed combination of netupitant and palonosetron: Review of clinical data. J Oncol Pharm Pract 2016;22:485–95.
26. Petri H. Das Interaktionspotenzial der oralen Antikoagulanzien. Krankenhauspharmazie 2020;41:267–72.
27. Sharma A, Hamelin BA. Classic histamine H1 receptor antagonists: a critical review of their metabolic and pharmacokinetic fate from a bird’s eye view. Curr Drug Metab. 2003;4:105–29.
28. Sharma A, Pibarot P, Pilote S, et al. Toward optimal treatment in women: the effect of sex on metoprolol-diphenhydramine interaction. J Clin Pharmacol 2010;50:214–25.
29. Verwimp-Hoeks MPA, van Herpen CML, Burger DM. Aprepitant quetiapine: a clinically significant drug interaction in a patient treated for head and neck cancer. Ann Oncol 2012;23:801–2.
30. Villikka K, Kivistö KT, Neuvonen PJ. The effect of rifampin on the pharmacokinetics of oral and intravenous ondansetron. Clin Pharmacol Ther 1999;65:377–81.
31. Vlase L, Leucuta A, Farcau D, et al. Pharmacokinetic interaction between fluoxetine and metoclopramide in healthy volunteers. Biopharm Drug Dispos 2006;27:285–9.
32. Yoshinari K, Ueda R, Kusano K, et al. Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem Pharmacol. 2008;76:139–45.
33. Yoshizato T, Kotegawa T, Imai H, et al. Itraconazole and domperidone: a placebo-controlled drug interaction study. Eur J Clin Pharmacol 2012;68:1287–94.
34. Zhou SF. Polymorphism of human cytochrome P4502D6 and its clinical significance: Part I. Clin Pharmacokinet. 2009;48:689–723.
*Nachdruck aus Krankenhauspharmazie 2021;42:21–5.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2021; 28(01):35-39