Holger Petri, Bad Wildungen*
Antiarrhythmika zur Rhythmuskontrolle
Klasse-I-Substanzen
Flecainid und Propafenon haben gegenüber anderen Arzneimitteln zur Rhythmuskontrolle von Vorhofflimmern ein günstiges Nebenwirkungsprofil und sollten bei Patienten ohne strukturelle Herzerkrankung bevorzugt verordnet werden [17]. Zudem sind sie als Pill-in-the-pocket-Medikation in der Anfallsbehandlung zugelassen [17]. Beide Wirkstoffe werden über das polymorph exprimierte Cytochrom-P450(CYP)-Isoenzym 2D6 verstoffwechselt. Der Anteil einer Dosis, der einer hepatischen Metabolisierung unterliegt, ist bei Flecainid gering, kann im Einzelfall von klinischer Bedeutung sein [43]. Die starken CYP2D6-Hemmer Chinidin und Paroxetin erhöhten in klinischen Studien den AUC(Fläche unter der Konzentrations-Zeit-Kurve)-Wert von Flecainid bei normaler CYP2D6-Stoffwechselaktivität durchschnittlich um 31 bzw. 28 % [32, 37]. In der Paroxetin-Studie war der Einfluss auf die Plasmaspiegel bei genetisch bedingter reduzierter CYP2D6-Metabolisierungskapazität nicht relevant [37].
In einer Kasuistik wird über eine ältere Patientin berichtet, die unter bestehender Paroxetin-Therapie innerhalb kurzer Zeit nach Beginn einer Flecainid-Einnahme mit 200 mg/d delirante Symptome entwickelt. Die Plasmaspiegel lagen über dem maximal empfohlenen Referenzbereich. Nach Halbierung der Flecainid-Dosis sistieren die Symptome nach drei Tagen. Neben der initial zu hohen Dosis hat die Abbauhemmung von Flecainid durch den starken CYP2D6-Inhibitor zu dem Ereignis beigetragen [44]. Es sollten bei Therapiebeginn und Dosisänderungen Kontrollen der Flecainid-Plasmaspiegel stattfinden [16]. Diese Empfehlung ist übertragbar bei Einleitung einer Therapie mit potenten CYP2D6-Hemmern. Die Bildung von CYP2D6-Enzymen kann durch die Komedikation nicht induziert werden (Abb. 1). Nach Herstellerangaben können aber Induktoren wie Carbamazepin die Flecainid-Spiegel um 30 % senken [16]. Eine mögliche Erklärung ist, dass bei Patienten mit genetisch bedingter reduzierter CYP2D6-Aktivität der metabolische Weg auch über CYP1A2 relevant werden kann, dessen Bildung induzierbar ist. So beschleunigt Zigarettenrauch den Abbau von Flecainid [27]. In einer weiteren Untersuchung zeigte sich bei Patienten mit reduzierter CYP2D6-Aktivität im höheren Alter eine langsamere Metabolisierungskapazität. Anders als bei CYP2D6-Substraten sinkt die Clearance von CYP1A2-Substraten mit dem Alter [4].
Dextromethorphan ist ein CYP2D6-Index-Substrat und es wird der Hauptmetabolit Dextrorphan gebildet. Flecainid erhöhte den metabolischen Dextromethorphan/Dextrorphan-Quotienten von 0,014 auf 0,163 [22].
Im Gegensatz zu Flecainid entsteht beim Abbau von Propafenon über CYP2D6 mit 5-Hydroxypropafenon ein aktiver Metabolit. In einer Studie mit neun Patienten, davon sieben mit Extensive- und zwei mit Poor-Metabolizer-Status, wurde der Einfluss von Chinidin auf die Pharmakokinetik von Propafenon untersucht. Bei den normalen Metabolisierern erhöhte sich die AUC durchschnittlich um das 2,7-Fache, die Spiegel des aktiven Metaboliten halbierten sich. Über CYP1A2 und CYP3A4 wird mit N-Depropylpropafenon ein zweiter aktiver Metabolit gebildet [14]. Sein Anstieg wird dadurch erklärt, dass durch die um mehr als 50%ige Blockade des Hauptabbauwegs die Metabolisierung vermehrt über den Seitenweg erfolgt. Der Metabolit trägt aufgrund seiner geringen Plasmakonzentration vernachlässigbar zur antiarrhythmischen Wirkung bei. Bei den beiden Poor Metabolizern änderten sich die Plasmaspiegel von Muttersubstanz und 5-Hydroxypropafenon nur unwesentlich und von N-Depropylpropafenon gar nicht [19]. Somit lässt sich festhalten, dass das Ausmaß der CYP2D6-Hemmung durch die Komedikation vom CYP2D6-Phänotyp abhängig ist. Wie sich CYP2D6-Inhibitoren bei Patienten vom ultraschnellen Metabolisierungs-Phänotyp auswirken, ist, wie bei Flecainid, nicht bekannt. Auch für Propafenon sollte folglich die Dosistitration sorgfältig erfolgen, wenngleich das therapeutische Fenster im Vergleich zu Flecainid mit 100–1500 ng/ml versus 200–700 ng/ml deutlich breiter ist [14, 16, 43].
Propafenon hemmt den Abbau von CYP2D6-Substraten. In einer klinischen Studie verdoppelte sich die Exposition einer Einzeldosis Metoprolol durch eine Einzeldosis Propafenon bei gesunden Probanden. Bei vier Patienten im Steady State stiegen die Betablocker-Spiegel durch Propafenon um das 2- bis 5-Fache [47]. Mit einer AUC-Steigerung von 54 % erhöhte Propafenon die Plasmaspiegel von Coffein, einem CYP1A2-Testsubstrat [36]. Bei Arzneistoffen mit enger therapeutischer Breite und Metabolismus über CYP1A2 wie Clozapin sollte dies beachtet werden.
Klasse-III-Substanzen
Amiodaron ist der Hauptvertreter diese Stoffklasse und das wirksamste Arzneimittel in der rhythmuserhaltenden Therapie bei Patienten mit Vorhofflimmern. Amiodaron hat jedoch relativ häufige nichtkardiale Nebenwirkungen, die regelmäßige Kontrolluntersuchungen erfordern [17, 35]. Bei Patienten mit schwerer Herzinsuffizienz zeigt Amiodaron ein akzeptables Sicherheitsprofil und bleibt als einziges rhythmuskontrollierendes Medikament [17, 35]. Zur Frequenzkontrolle dient Amiodaron nur als Reserve, wenn andere Medikamentenklassen aufgrund von Begleiterkrankungen nicht genommen werden können oder diese nicht genügend wirksam sind [17]. Amiodaron birgt ein komplexes pharmakokinetisches Interaktionsprofil als „Victim Drug“ und als „Perpetrator“ [33]. Durch N-Dealkylierung über CYP3A4 und nachgeordnet CYP2C8 entsteht der aktive Metabolit Desethylamiodaron [8, 29].
In einer klinischen Studie erhöhte Grapefruitsaft durch CYP3A4-Hemmung die Exposition einer Einzeldosis Amiodaron um 50 %. Die Bildung des aktiven Metaboliten N-Desethylamiodaron wurde nahezu komplett unterbunden [31]. Dies gibt einen Hinweis, wie sich starke CYP3A4-Inhibitoren auf die Amiodaron-Exposition auswirken können. In verschiedenen Kasuistiken werden Torsade-de-pointes(TdP)-Arrhythmien bei Kombination mit dem starken CYP3A4-Inhibitor Clarithromycin beschrieben. Das Makrolid-Antibiotikum hat, wie andere rhythmuserhaltende Substanzen, selbst ein hohes TdP-Potenzial [50]. Eine Kombination aus pharmakodynamischen und -kinetischen Wechselwirkungen führte zu den unerwünschten kardialen Ereignissen [46]. Wie sich CYP2C8-Hemmer auf die Amiodaron-Exposition auswirken, ist nicht bekannt. Die klinische Bedeutung ist vermutlich nicht ausgeprägt, da, wie am Beispiel von Grapefruitsaft gezeigt, nach Blockade über CYP3A4 keine nennenswerten Desethylamiodaron-Spiegel gemessen wurden.
Induktoren beschleunigen den Abbau von Amiodaron und können so ohne Dosiserhöhung zu subtherapeutischen Konzentrationen führen.
In einem Fallbericht wurde gezeigt, wie die Plasmaspiegel von Amiodaron unter Rifampicin-Therapie durch therapeutisches Drug-Monitoring (TDM) im Zielbereich gehalten werden können. Während der Antibiose wird hierzu zeitweise eine bis zu 4,5-fache Dosiserhöhung des Antiarrhythmikums benötigt. Der metabolische Quotient Metabolit zu Muttersubstanz kehrt sich von 0,8 auf 1,8 um und fällt nach Ende der Rifampicin-Einnahme wieder auf den Ausgangswert [38].
Die Vortexkeratopathie, eine wirbelartige Ablagerung im Hornhautepithel des Auges, ist eine typische unerwünschte Arzneimittelwirkung einer langfristigen Amiodaron-Anwendung. Bei einer Patientin verschwindet diese Nebenwirkung, nachdem wegen einer Tuberkulose eine Rifampicin-Therapie eingeleitet wurde. Die gemessenen Plasmaspiegel lagen unterhalb des therapeutischen Bereichs. Nach Ende der Antibiose stiegen die Plasmaspiegel wieder und die Vortexkeratopathie kehrte zurück [34].
Amiodaron und seine Metaboliten haben das Potenzial, CYP 1A2, 2C9, 2D6 und 3A4 zu hemmen [33]. Die Steigerung der AUC fällt beim CYP1A2-Substrat Lidocain mit 21 % gering aus. Warfarin besteht als racemisches Gemisch zu gleichen Teilen aus S- und R-Warfarin. S-Warfarin besitzt eine 2- bis 5-fach höhere blutgerinnungshemmende Aktivität als R-Warfarin. S-Warfarin wird primär über CYP2C9 abgebaut [39]. Amiodaron führte in einer Studie zu einer 2,1-fachen Erhöhung der AUC von S-Warfarin [33]. In einer schwedischen Untersuchung erhöhte sich der INR-Wert durchschnittlich von 2,6 auf 3,1. 37 % der Patienten hatten nach Beginn einer Amiodaron-Therapie einen INR-Wert > 3,0 [26]. Die Einnahme von Amiodaron führte bei Patienten zu einer Verdoppelung der Metoprolol-Exposition, wobei das Ausmaß vom CYP2D6-Genotyp abhängig war. Bei Trägern von zwei oder mehr funktionsfähigen Allelen stiegen die Spiegel um das 2,3-Fache, bei denen mit einem funktionsfähigen Allel nur um 61 % [49]. Amiodaron erhöhte bei gesunden Probanden den AUC-Wert des CYP3A4-Substrats Simvastatin um 73 % [33]. Sieben Fälle von Rhabdomyolysen bei Patienten mit der Kombination Amiodaron und Simvastatin (80 mg/d) wurden in der Search-Studie beobachtet, jedoch kein Fall unter Komedikation mit Simvastatin in einer Dosis von 20 mg/d. In der Folge wurde die Dosis des Statins bei Kombination mit Amiodaron auf 20 mg am Tag begrenzt [1].
Dronedaron kann bei koronarer Herzkrankheit, relevanter Herzklappenerkrankung oder abnormer linksventrikulärer Hypertrophie bei Abwesenheit einer signifikanten Herzinsuffizienz zum Einsatz kommen [17]. Dronedaron ist ein CYP3A4-Substrat. Es entsteht ein aktiver Metabolit, dessen pharmakodynamische Aktivität aber um das 3- bis 10-Fache geringer wirksam ist als die der Muttersubstanz. Die wiederholte Gabe des starken CYP3A4-Inhibitors Ketoconazol ergab eine 17-fache Zunahme der Exposition. Somit ist eine gleichzeitige Einnahme von starken CYP3A4-Inhibitoren (Abb. 1) mit Dronedaron kontraindiziert. Der vergleichsweise weniger starke CYP3A4-Hemmer Erythromycin führte zu einer Steigerung der AUC um das 3,8-Fache. In einem ähnlichen Ausmaß erhöhte wiederholter Konsum von Grapefruitsaft die Dronedaron-Exposition. Diltiazem und Verapamil vermochten die Plasmaspiegel von Dronedaron um 70 bzw. 40 % zu erhöhen.
Rifampicin als starker CYP3A4-Induktor (Abb. 1) senkte die Dronedaron-Exposition um 80 % ohne Veränderung der Exposition des aktiven Metaboliten. Dronedaron sollte mit starken CYP3A4-Induktoren nicht kombiniert werden. Dronedaron kann über CYP3A4-Hemmung und Inhibition des Effluxtransporters P-Glykoprotein (P-gp) die Exposition anderer Arzneimittel beeinflussen. So erhöhte Dronedaron die Plasmaspiegel von Simvastatinsäure, des aktiven Metaboliten von Simvastatin, um das Doppelte und die von Atorvastatin, ebenfalls ein CYP3A4-Substrat, um das 1,7-Fache. Die Plasmaspiegel von Dabigatranetexilat, einem P-gp-Substrat, verdoppelten sich durch Dronedaron. Eine Kombination ist daher kontraindiziert. Eine Komedikation mit Digoxin erhöhte über P-gp-Hemmung dessen Exposition um das 2,5-Fache, was eine Dosisreduktion um 50 % erfordert [12].
Sotalol zählt zu den nichtselektiven Betablockern, hemmt aber auch Kaliumkanäle und hat von den rhythmuskontrollierenden Antiarrhythmika die größte proarrhythmische Wirkung mit einem vergleichsweise höheren Risiko für TdP-Arrhythmien [17, 35]. Sotalol wird hauptsächlich renal unverändert ausgeschieden [15].
Antiarrhythmika zur Frequenzkontrolle
Klasse-II-Substanzen
Antiarrhythmika der Klasse II sind Arzneistoffe mit Beta-Rezeptoren-blockierender Wirkung. Die meistverwendeten Wirkstoffe sind Bisoprolol, Carvedilol, Metoprolol und Nebivolol [17].
Bisoprolol wird zu 50 % in der Leber zu inaktiven Metaboliten abgebaut, die verbleibenden 50 % werden renal eliminiert. Am metabolischen Abbau ist CYP3A4 beteiligt, jedoch sind keine relevanten Interaktionen mit CYP3A4-Hemmern beschrieben. Die Enzyminduktion von CYP3A4 durch Rifampicin führt zu einer Senkung der AUC um ein Drittel. Dies erfordert in der Regel keine Dosisanpassung [28]. In einem Fallbericht kommt es bei einem seit 15 Jahren mit Bisoprolol gut eingestellten Patienten zu einem manifesten Blutdruckanstieg und erstmals auftretenden Herzrhythmusstörungen. Aufgrund einer Tuberkulose-Infektion wurde Rifampicin neu angesetzt. In der Folge sank die Bisoprolol-Exposition um ein Drittel. Die Dosierung von Bisoprolol wurde auf 3,75 mg morgens und zusätzlich 1,875 mg abends umgestellt. Durch diese Dosiserhöhung konnte der erhöhte Blutdruck auf Normwerte gesenkt werden [5].
Carvedilol ist ein nichtselektiver Betablocker mit Alpha-1-Rezeptoren-blockierenden Eigenschaften. Carvedilol ist ein Razemat. Beide Isomere wirken als Alphablocker, wogegen primär das S-Enantiomer für die Blockierung der Beta-Rezeptoren verantwortlich ist. Der oxidative Metabolismus von Carvedilol ist stereoselektiv. Das R-Enantiomer wird überwiegend durch CYP2D6 metabolisiert, das S-Enantiomer zusätzlich über CYP2C9 [9]. Fluoxetin als starker CYP2D6-Inhibitor führte bei Patienten mit Herzinsuffizienz im Steady State zu einer Erhöhung der mittleren AUC des R-Enantiomers um 77 % und einer statistisch nicht signifikanten Erhöhung des S-Enantiomers [21]. Bei zwölf gesunden Probanden stieg die Exposition einer Einzeldosis von Carvedilol durch Paroxetin. Das Antidepressivum erhöhte die AUC des R-Enantiomers um das 2,5- und des S-Enantiomers um das 1,9-Fache. Klinische Parameter wie Blutdruck und Herzrate blieben aber bei den Probanden ohne klinisch relevante Effekte [42]. Amiodaron erhöhte bei Patienten mit Herzinsuffizienz die AUC des S-Enantiomers über Hemmung von CYP2C9 um das 2,2-Fache, während es zu keinem signifikanten Anstieg des R-Isomers kam [18]. Bei einem nierentransplantierten Patienten führte die Verordnung des starken CYP2C9-Hemmers Fluconazol zu der bestehenden Medikation aus Carvedilol und Tacrolimus innerhalb weniger Tage zu einer dekompensierten Herzfunktion, die sich in einer ausgeprägten Bradykardie und Blutdruckabfall äußerte. Die erhöhten S-Carvedilol-Plasmaspiegel sind durch diese Interaktion mitverantwortlich für die unerwünschte Arzneimittelwirkung [41].
Metoprolol wird in der Leber vorwiegend oxidativ über CYP2D6 metabolisiert. Die Hauptmetaboliten zeigen nur schwach Betarezeptoren-blockierende Wirkungen [6]. In zahlreichen Studien wurden die Plasmaspiegelerhöhungen von Metoprolol durch CYP2D6-Inhibitoren auch in Abhängigkeit des Metabolisiererstatus evaluiert. Dabei zeigte sich, dass die Plasmaspiegel weniger ansteigen bei Patienten bzw. Probanden mit einem Poor-Metabolizer(PM)-Status verglichen mit extensive Metabolizern (EM). Wie bei Amiodaron war der Anstieg der AUC um 103 % bei Trägern von zwei funktionsfähigen Allelen in Kombination mit Celecoxib höher als bei denen mit nur einem funktionsfähigen Allel mit 36 % [48]. Bei Diphenhydramin lag der Anstieg bei Personen mit EM-Phänotyp bei 61 %, bei PM-Personen lediglich bei 10 % [23]. Eine früher angenommene relevante CYP2D6-Hemmung durch Metoprolol konnte in einer Studie widerlegt werden [25].
Nebivolol ist ein hochselektiver Betablocker mit zusätzlich leicht vasodilatierenden Eigenschaften aufgrund einer NO-Freisetzung aus Endothelzellen [13, 17]. Nebivolol wird umfassend verstoffwechselt und es entsteht über CYP2D6 ein aktiver Hydroxymetabolit. Die orale Bioverfügbarkeit variiert in Abhängigkeit des CYP2D6-Phänotyps. Bei schnellen CYP2D6-Metabolisierern beträgt sie 12 %, bei langsamen ist sie nahezu vollständig. Maximale Plasmakonzentrationen der Muttersubstanz sind bei langsamen Metabolisierern im Steady State 10- bis 23-mal höher als bei extensiven Metabolisierern. Jedoch ist die Summe der maximalen Plasmakonzentration der aktiven Fraktion bei Poor-Metabolizern nur wenig höher als bei Extensive-Metabolizern [13, 30]. Klinisch zeigte sich bei 37 Patienten, von denen 14 Poor- und 23-Extensive-Metabolizer waren, trotz der zum Teil deutlichen Veränderungen pharmakokinetischer Parameter keine klinisch relevanten Unterschieden in der antihypertensiven Wirkung und Verträglichkeit. Jedoch war die Frequenzsenkung bei den EM-Patienten höher als bei den PM-Patienten [30]. In zwei klinischen pharmakokinetischen Interaktionsstudien mit gesunden Probanden wurden die AUC-Veränderungen und klinische Folgen einer Einzeldosis Nebivolol in Kombination mit den starken CYP2D6-Inhibitoren Bupropion und Paroxetin untersucht. Obwohl die Exposition der aktiven Fraktion jeweils deutlich angestiegen ist, blieb das pharmakodynamische Ansprechen auf Blutdruck und Herzrate unbeeinflusst. Es besteht weiterer Forschungsbedarf nach den klinischen Konsequenzen einer Abbauhemmung von Nebivolol im Steady State [3, 20]. Auch gibt es keine Untersuchungen, wie sich CYP2D6-Hemmer bei Patienten mit einem UM-Phänotyp auswirken. Nach Herstellerangaben wird aufgrund der Unterschiede bei den Metaboliserungsraten eine Dosierung von Nebivolol entsprechend der individuellen Erfordernisse empfohlen [13]. Dies sollte ebenso bei Anwendung mit potenten CYP2D6-Inhibitoren gelten.
Klasse-IV-Substanzen
Diltiazem und Verapamil eignen sich besonders dann zur Frequenzkontrolle, wenn eine Betablocker-Therapie aufgrund von Kontraindikationen, v. a. einer unzureichend kontrollierten obstruktiven Atemwegserkrankung nicht anwendbar ist [17]. Diltiazem hemmt als CYP3A4-Inhibitor seinen eigenen Metabolismus. Durch starke CYP3A4-Inhibitoren wurde eine mäßige Erhöhung um weniger als das 2-Fache der Plasmaspiegel von Diltiazem beschrieben [10]. Die Kombination mit dem CYP3A4-Induktor Rifampicin führt zu einem beschleunigten Abbau, sodass Diltiazem nicht mehr nachweisbar ist [40]. Inwiefern die schwächer wirksamen Metaboliten diesen beschleunigten Abbau kompensieren ist nicht bekannt. Als CYP3A4-Hemmer erhöhte Diltiazem die Exposition der CYP3A4-Testsubstrate Midazolam und Triazolam um das 3,8- und 3,0-Fache [2, 45].
Verapamil wird wie Diltiazem primär über CYP3A4 metabolisiert. Von den zwölf Metaboliten wird allein Norverapamil eine gewisse pharmakologische Wirkung zugesprochen. Grapefruitsaft erhöhte die AUC der beiden Enantiomere R- und S-Verapamil um 49 bzw. 37 %. Stärkere CYP3A4-Inhibitoren sollten die Exposition in einem größeren Ausmaß steigern, wobei dann der Metabolismus auch über andere Cytochrom-Isoenzyme stattfinden könnte. Induktoren beschleunigen den oxidativen Metabolismus von Verapamil. So erhöhte Phenobarbital die Clearance um das 5-Fache. Rifampicin senkte die AUC bei oraler Gabe von Verapamil um 92 % [11]. Es ist mit einem Wirkungsverlust und damit einer Wiederkehr von zum Beispiel supraventrikulärer Arrhythmien zu rechnen.
Wie Diltiazem kann Verapamil die Exposition von CYP3A4-Substraten über eine Hemmung des Metabolismus steigern. Verapamil erhöhte den AUC-Wert von oral eingenommenem Midazolam um das 2,9-Fache [2]. In einer ähnlichen Größenordnung lagen die Steigerungen von anderen CYP3A4-Substraten wie Everolimus, Sirolimus und Simvastatin. Zusätzliche Interaktionsrisiken ergeben sich im Vergleich zu Diltiazem durch Hemmung von P-Glykoprotein. Die Bioverfügbarkeit des DOAK Dabigatranetexilat erhöhte sich bei zeitgleicher Einnahme von schnell freisetzendem Verapamil um 50 % [11].
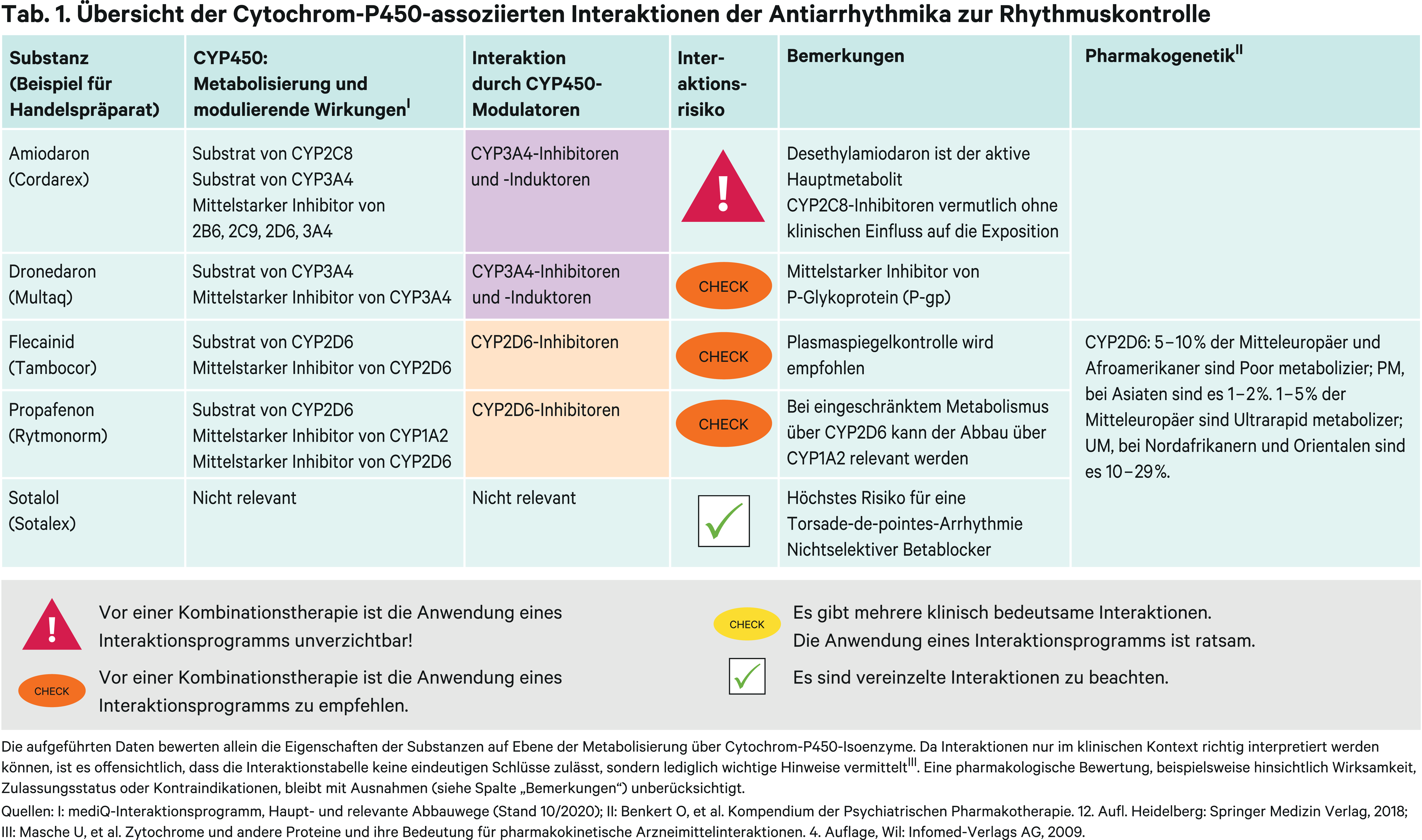
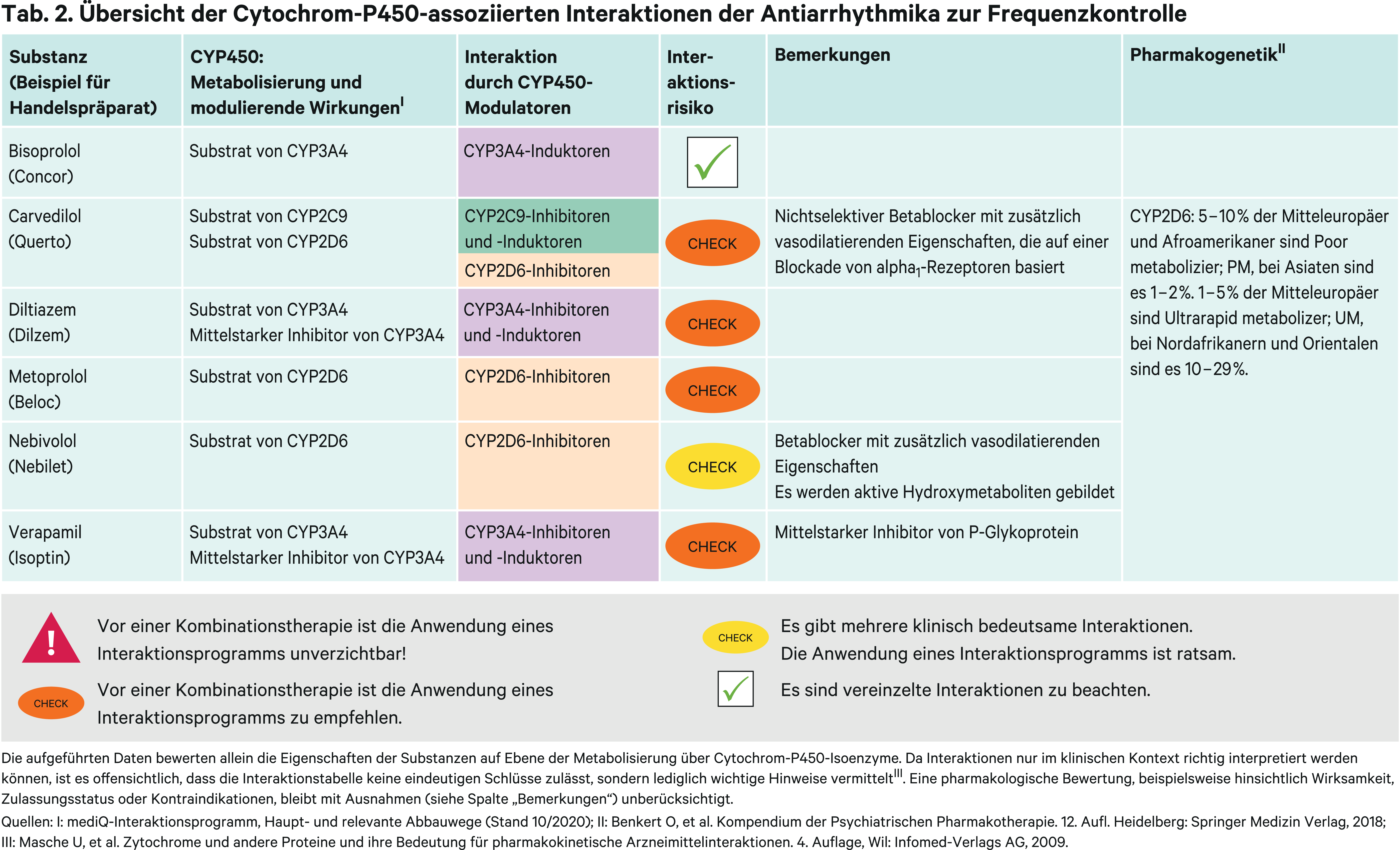
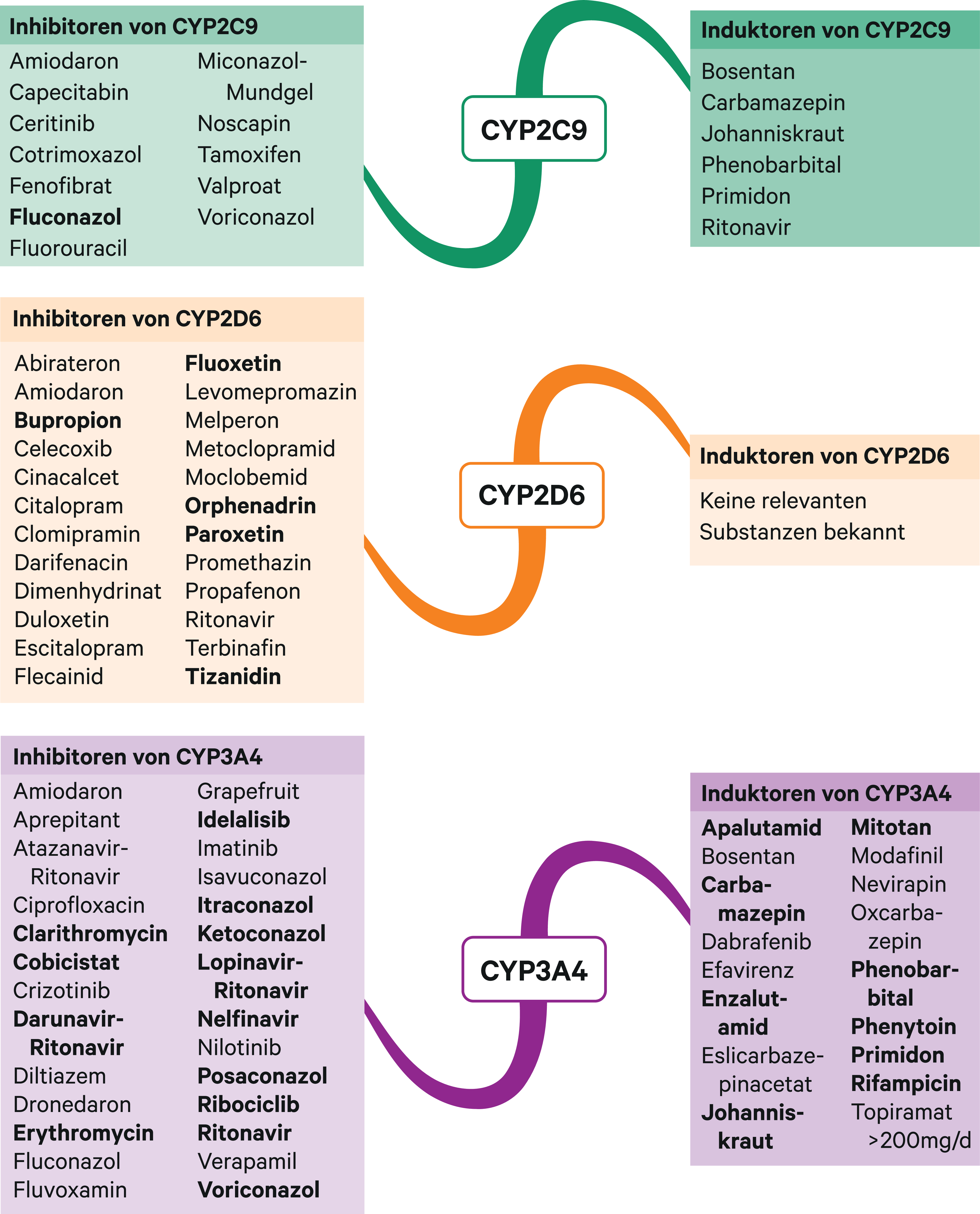
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2C9, 2D6 und 3A4 (Stand 10/2020) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Arzneimittelkommission der deutschen Ärzteschaft. Rhabdomyolysen durch Interaktion von Simvastatin und Amiodaron. Drug Safety Mail 2017–06.
2. Backman JT, Olkkola KT, Aranko K, et al. Dose of midazolam should be reduced during diltiazem and verapamil treatments. Br J Clin Pharmacol 1994;37:221–5.
3. Briciu C, Neag M, Muntean D, et al. A pharmacokinetic drug interaction study between nebivolol and paroxetine in healthy volunteers. J Clin Pharm Ther 2014;39:535–40.
4. Doki K, Homma M, Kuga K, et al. Effects of CYP2D6 genotypes on age-related change of flecainide metabolism: involvement of CYP1A2-mediated metabolism.Br J Clin Pharmacol. 2009;68:89–96.
5. Drechsler A, Kirch W. Tuberkulose durch Bacillus-Calmette-Guérin-Instillation bei Blasenkarzinom und Hypertonie. Verminderte Bisoprolol-Wirkung durch Rifampicin. Dtsch Med Wochenschr 2010;135:1968–70.
6. Fachinformation Beloc-Zok®. Stand: April 2018.
7. Fachinformation Concor®. Stand: Februar 2020.
8. Fachinformation Cordarex®. Stand: Mai 2020.
9. Fachinformation Dilatrend®. Stand: Januar 2020.
10. Fachinformation Dilzem®. Stand: April 2018.
11. Fachinformation Isoptin®. Stand: Januar 2020.
12. Fachinformation Multaq®. Stand: September 2019.
13. Fachinformation Nebilet®. Stand: August 2018.
14. Fachinformation Rytmonorm®. Stand: April 2019.
15. Fachinformation Sotalex®. Stand: Juli 2019.
16. Fachinformation Tambocor®. Stand: Juli 2017.
17. Fischer TH. Antiarrhythmika: Welche Rolle spielen sie heute noch? Dtsch Med Wochenschr 2020;145:526–35.
18. Fukumoto K, Kobayashi T, Komamura K, et al. Stereoselective effect of amiodarone on the pharmacokinetics of racemic carvedilol. Drug Metab Pharmacokinet 2005;20:423–7.
19. Funck-Brentano C, Kroemer HK, Pavlou H, et al. Genetically-determined interaction between propafenone and low dose quinidine: role of active metabolites in modulating net drug effect. Br J Clin Pharmacol 1989;27:435–44.
20. Gheldiu AM, Popa A, Neag M, et al. Assessment of a Potential Pharmacokinetic Interaction between Nebivolol and Bupropion in Healthy Volunteers. Pharmacology 2016;98:190–8.
21. Graff DW, Williamson KM, Pieper JA, et al. Effect of fluoxetine on carvedilol pharmacokinetics, CYP2D6 activity, and autonomic balance in heart failure patients. J Clin Pharmacol 2001;41:97–106.
22. Haefeli WE, Bargetzi MJ, Follath F, et al. Potent inhibition of cytochrome P450IID6 (debrisoquin 4-hydroxylase) by flecainide in vitro and in vivo. J Cardiovasc Pharmacol 1990;15:776–9.
23. Hamelin BA, Bouayad A, Méthot J, et al. Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther 2000;67:466–77.
24. Hanke N, Türk D, Selzer D, et al. A Mechanistic, Enantioselective, Physiologically Based Pharmacokinetic Model of Verapamil and Norverapamil, Built and Evaluated for Drug-Drug Interaction Studies. Pharmaceutics 2020;12:556.
25. Hefner G, Unterecker S, Shams ME, et al. Melperone but not bisoprolol or metoprolol is a clinically relevant inhibitor of CYP2D6: evidence from a therapeutic drug monitoring survey. J Neural Transm (Vienna) 2015;122:1609–17.
26. Holm J, Lindh JD, Andersson ML, Mannheimer B. The effect of amiodarone on warfarin anticoagulation: a register-based nationwide cohort study involving the Swedish population. J Thromb Haemost 2017;15:446–53.
27. Holtzman JL, Weeks CE, Kvam DC, et al. Identification of drug interactions by meta-analysis of premarketing trials: the effect of smoking on the pharmacokinetics and dosage requirements for flecainide acetate. Clin Pharmacol Ther 1989;46:1–8.
28. Kirch W, Rose I, Klingmann I, et al. Interaction of bisoprolol with cimetidine and rifampicin. Eur J Clin Pharmacol 1986;31:59–62.
29. Klotz U. Antiarrhythmics: elimination and dosage considerations in hepatic impairment. Clin Pharmacokinet 2007;46:985–96.
30. Lefebvre J, Poirier L, Poirier P, et al. The influence of CYP2D6 phenotype on the clinical response of nebivolol in patients with essential hypertension. Br J Clin Pharmacol 2007;63:575–82.
31. Libersa CC, Brique SA, Motte KB, et al. Dramatic inhibition of amiodarone metabolism induced by grapefruit juice. Br J Clin Pharmacol. 2000;49:373–78.
32. Lim KS, Cho JY, Jang IJ, et al. Pharmacokinetic interaction of flecainide and paroxetine in relation to the CYP2D6*10 allele in healthy Korean subjects. Br J Clin Pharmacol 2008;66:660–6.
33. McDonald MG, Au NT, Rettie AE. P450-Based Drug-Drug Interactions of Amiodarone and its Metabolites: Diversity of Inhibitory Mechanisms. Drug Metab Dispos 2015;43:1661–69.
34. Mehta S, Bunya VY, Orlin SE, et al. A significant drug-drug interaction detected through corneal examination: resolution of cornea verticillata while using amiodarone. Cornea. 2012;31:81–3.
35. Metzner A, Rillig A, Kirchhof P. Medikamente oder Ablation. Perspektiven der Kardiologie 1/2020, Dtsch Ärzteblatt 14–7.
36. Michaud V, Mouksassi MS, Labbé L, et al. Inhibitory effects of propafenone on the pharmacokinetics of caffeine in humans. Ther Drug Monit 2006;28:779–83.
37. Munafo A, Buclin T, Tuto D, Biollaz J. The effect of a low dose of quinidine on the disposition of flecainide in healthy volunteers. Eur J Clin Pharmacol. 1992;43:441–3.
38. Oude Munnink TH, Demmer A, Slenter RHJ, et al. Amiodarone Rifampicin Drug-Drug Interaction Management With Therapeutic Drug Monitoring. Ther Drug Monit 2018;40:159–61.
39. Petri H. Das Interaktionspotenzial der oralen Antikoagulanzien. Krankenhauspharmazie 2020;41:267–72.
40. Prescribing information Cardiazem®. Stand November 2014.
41. Saliaj M, Taweesedt P; Chang M, et al. Fluconazole: A culprit in augmenting the risk of bradycardia leading to cardiac arrest. Critical Care Medicine 47 (Suppl. 1):62:Abstr. 161, Jan 2019.
42. Stout SM, Nielsen J, Bleske BE, et al. The impact of paroxetine coadministration on stereospecific carvedilol pharmacokinetics. J Cardiovasc Pharmacol Ther 2010;15:373–9.
43. Tamargo J, Le Heuzey J, Mabo P. Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide. Eur J Clin Pharmacol 2015;71:549–67.
44. Tsao YY, Gugger JJ. Delirium in a patient with toxic flecainide plasma concentrations: the role of a pharmacokinetic drug interaction with paroxetine. Ann Pharmacother 2009;43:1366–9.
45. Varhe A, Olkkola KT, Neuvonen PJ. Diltiazem enhances the effects of triazolam by inhibiting its metabolism. Clin Pharmacol Ther 1996;59:369–75.
46. Vieweg WV, Hancox JC, Hasnain M, et al. Clarithromycin, QTc interval prolongation and torsades de pointes: the need to study case reports. Ther Adv Infect Dis 2013;1:121–38.
47. Wagner F, Kalusche D, Trenk D, et al. Drug interaction between propafenone and metoprolol. Br J Clin Pharmacol 1987;24:213–20.
48. Werner U, Werner D, Rau T, et al. Celecoxib inhibits metabolism of cytochrome P450 2D6 substrate metoprolol in humans. Clin Pharmacol Ther 2003;74:130–7.
49. Werner D, Wuttke H, Fromm MF, et al. Effect of amiodarone on the plasma levels of metoprolol. Am J Cardiol 2004;94:1319–21.
50. Woosley RL, Heise CW, Gallo T, et al. www.CredibleMeds.org (letzter Zugriff am 4. Oktober 2020).
*Nachdruck aus Krankenhauspharmazie 2020;41:436–43.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, Prof. Dr. Peter Trenkwalder, Starnberg und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2020; 27(06):306-313