Holger Petri, Bad Wildungen*
Acetylsalicylsäure
Acetylsalicylsäure (ASS) hemmt über Acetylierung die Cyclooxygenase 1 (COX-1) irreversibel, wodurch der starke Plättchenaktivator Thromboxan A2 nicht mehr gebildet wird [3].
Metaboliten von ASS in niedriger Dosis wie Salicylsäure und seine Konjugationsprodukte haben keine Bedeutung für die Plättchen-hemmende Wirkung. Relevante CYP-bedingte Interaktionen sind nicht zu erwarten.
ADP-Rezeptor-Antagonisten (P2Y12-Inhibitoren)
Zu den ADP-Rezeptor-Antagonisten zählen die Thienopyridine Clopidogrel, Prasugrel und Ticlopidin sowie Ticagrelor und Cangrelor. Die Substanzen blockieren selektiv die Bindung von Adenosindiphosphat (ADP) an seine Thrombozytenrezeptoren, den Purinrezeptoren P2Y12. In der Folge unterbleibt die ADP-induzierte Thrombozytenaktivierung [13].
Clopidogrel, Prasugrel und Ticlopidin hemmen irreversibel die ADP-Rezeptoren. Sie sind Prodrugs, das heißt, die pharmakologisch aktiven Metaboliten entstehen durch enzymatische Umwandlung [13].
Thienopyridine
Clopidogrel ist der Hauptvertreter der ADP-Rezeptor-Antagonisten [20]. Etwa 15 % der Clopidogrel-Dosis werden über zwei Schritte bioaktiviert (Abb. 1) [13, 17]. Beteiligt sind verschiedene Cytochrom-P450(CYP)-Isoenzyme, wovon dem polymorph exprimierten CYP2C19 eine vorherrschende Rolle zugesprochen wird [7, 11–13, 16]. Zwei Polymorphismen sind besonders relevant. Patienten mit einem Poor-Metabolizer-Status (homozygote Träger des *2-Allels) bilden geringere Mengen an aktiven Metaboliten. Ultrarapid-Metabolisierer (*1/*17-, *17/*17-Allelträger) haben eine höhere Metabolisierungskapazität und es kommt dadurch zu einer gesteigerten Aktivierung von Clopidogrel [12, 21]. Beides kann zu einem veränderten Ansprechen beitragen. Im einen Fall steigt das Risiko für thrombotische Ereignisse, im anderen das für Blutungen [12]. Der Protonenpumpeninhibitor (PPI) Omeprazol und sein S-Enantiomer Esomeprazol sind CYP2C19-Inhibitoren (Abb. 2). Omeprazol in der Höchstdosis von 80 mg/Tag verminderte die Exposition gegenüber dem aktiven Metaboliten um 40 % in der Erhaltungsdosis. Hierdurch sank die Thrombozytenaggegationshemmung um 21 % [7]. Die klinische Bedeutung der Wechselwirkung mit Omeprazol und Esomeprazol ist noch nicht abschließend geklärt. Als alternativer PPI wird Pantoprazol empfohlen. Andere potente CYP2C19-Inhibitoren wie Fluconazol und Fluoxetin sollten ebenfalls vermieden werden [7]. Bei Einnahme von Induktoren kann analog der Empfehlung bei Ultrarapid-Metabolisierern die gemäß Zulassung empfohlene Dosis verordnet werden [21].
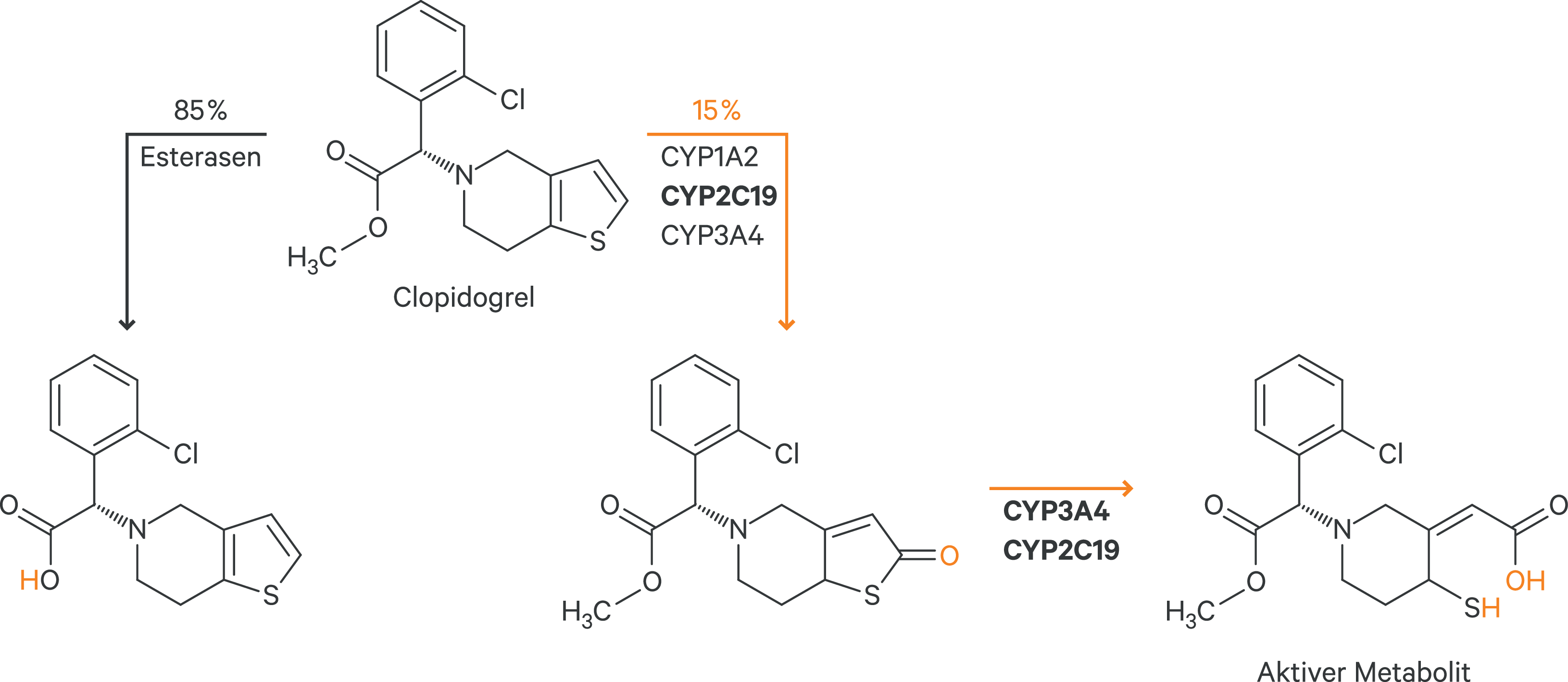
Abb. 1. Metabolismus von Clopidogrel (mod. nach [13] und https://www.pharmgkb.org/ [Stand: 04/2020])
Eine Ursache der nicht konsistenten Datenlage ist, dass andere Isoenzyme wie CYP1A2 und CYP3A4 eine relevante Beteiligung an der Bildung des Thiolmetaboliten haben. So hatten rauchende Patienten höhere Metabolitspiegel als Nichtraucher [14]. Polyzyklische Kohlenwasserstoffe im Zigrarettenrauch induzieren die Bildung von CYP1A2 [18].
CYP3A4 hat gemäß In-vitro-Untersuchungen besonders am zweiten Schritt des Metabolismus einen großen Anteil [17, 26]. Dies bestätigte sich durch In-vivo-Untersuchungen. So senkte der CYP3A4-Inhibitor Ketoconazol den Wert der 24h-AUC (Fläche unter der Konzentrations-Zeit-Kurve) des aktiven Metaboliten in der 75-mg-Erhaltungsdosis um 29 % [10]. Cobicistat und Ritonavir sind als starke CYP3A4-Hemmer pharmakokinetische Wirkverstärker (Booster) antiretroviraler Arzneimittel [19]. Ritonavir reduzierte in einer Studie mit zwölf gesunden Probanden den AUC0-4h-Wert um die Hälfte [15]. HIV-Patienten hatten in einer anderen Untersuchung knapp 70 % niedrigere Metabolitspiegel als die Vergleichsgruppe [19]. Es wird von der gleichzeitigen Anwendung von Clopidogrel mit geboosterten antiretroviralen Therapien (ART) abgeraten [7].
Clopidogrel hemmt CYP2B6 [22]. Wechselwirkungen mit CYP2B6-Substraten wie Cyclosphosphamid, Efavirenz und Selegilin sind daher möglich.
Ein Glucuronidmetabolit von Clopidogrel zeigte in einer Studie mit gesunden Probanden ein CYP2C8-hemmendes Potenzial. Die Plasmaspiegel des CYP2C8-Testsubstrats Repaglinid stiegen bei der 300-mg-Dosis um das 5-Fache und bei der 75-mg-Dosis um das 4-Fache [25]. Somit gilt sowohl besondere Vorsicht bei der Kombination von Clopidogrel mit Repaglinid als auch mit anderen CYP2C8-Substraten wie Dasabuvir und Paclitaxel [7, 15].
Prasugrel wird über CYP3A4 und 2B6 zum aktiven Metaboliten verstoffwechselt [5]. Während Ketoconazol die Metabolitexposition nicht veränderte, reduzierten Ritonavir/Cobicistat-geboosterte ART die Metabolitbildung um die Hälfte. Dies war jedoch ohne Einfluss auf die Thrombozytenaggegationshemmung, sodass Prasugrel hier eine Alternative zu Clopidogrel darstellt [1, 5, 10, 19].
Rifampicin hatte keinen signifikanten Einfluss auf die Pharmakokinetik von Prasugrel [5, 9]. Der Effekt von CYP3A4-Induktoren auf die Bildung des aktiven Metaboliten kann als klinisch nicht relevant angesehen werden [5].
Der Mechanismus der Bioaktivierung von Ticlopidin sowie die beteiligten Enzyme sind nicht vollständig geklärt [13]. Es kann aber angenommen werden, dass CYP-Enzyme eine Rolle spielen könnten [2]. Ticlopidin ist ein starker CYP2B6-Inhibitor [8].
Direkt wirkende P2Y12-Inhibitoren
Ticagrelor und Cangrelor (i. v.) sind direkte und reversible Inhibitoren der Thrombozytenaggregation [13].
Ticagrelor ist ein Substrat von CYP3A4. Der Hauptmetabolit ist ebenso thrombozytenaggregationshemmend wie die Muttersubstanz und macht etwa 30 bis 40 % der Plasmaskonzentration von Ticagrelor aus [4]. In einer Arbeit wurde der Einfluss von Ketoconazol und des mittelstarken CYP3A4-Inhibitors Diltiazem auf die AUC-Werte von Muttersubstanz und aktiven Metaboliten untersucht. Ketoconazol erhöhte die Exposition von Ticagrelor um das 7,3-Fache und senkte die des Metaboliten um 56 %. Diltiazem steigerte den AUC-Wert um das 2,7-Fache, die Konzentration des Metaboliten blieb unverändert [23]. Starke CYP3A4-Hemmer sind folglich unter einer Ticagrelor-Therapie kontraindiziert. Der Einfluss durch Diltiazem wird bei den meisten Patienten als nicht klinisch relevant betrachtet und andere mittelstarke CYP3A4-Hemmer (Abb. 2) können zusammen mit Ticagrelor verordnet werden [4].
Rifampicin reduzierte den AUC-Wert einer Ticagrelor-Einzeldosis um 86 % und den des Metaboliten um 46 % [24]. Starke CYP3A4-Induktoren sollten nicht mit Ticagrelor kombiniert werden [4].
Der Cangrelor-Metabolismus ist CYP-unabhängig und CYP-Isoenzyme werden von therapeutischen Cangrelor-Konzentrationen oder seinen wesentlichen Metaboliten nicht gehemmt [6].
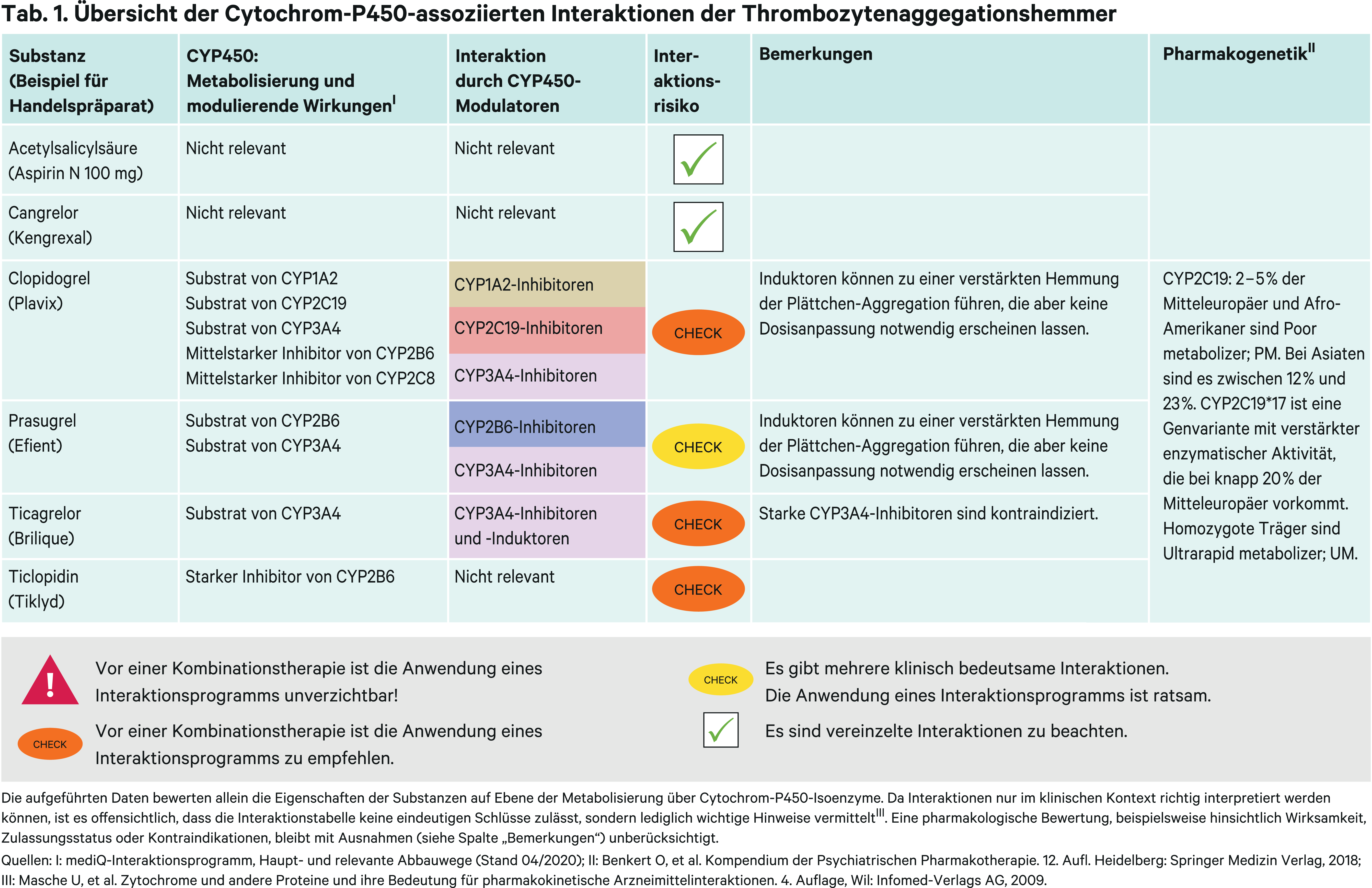
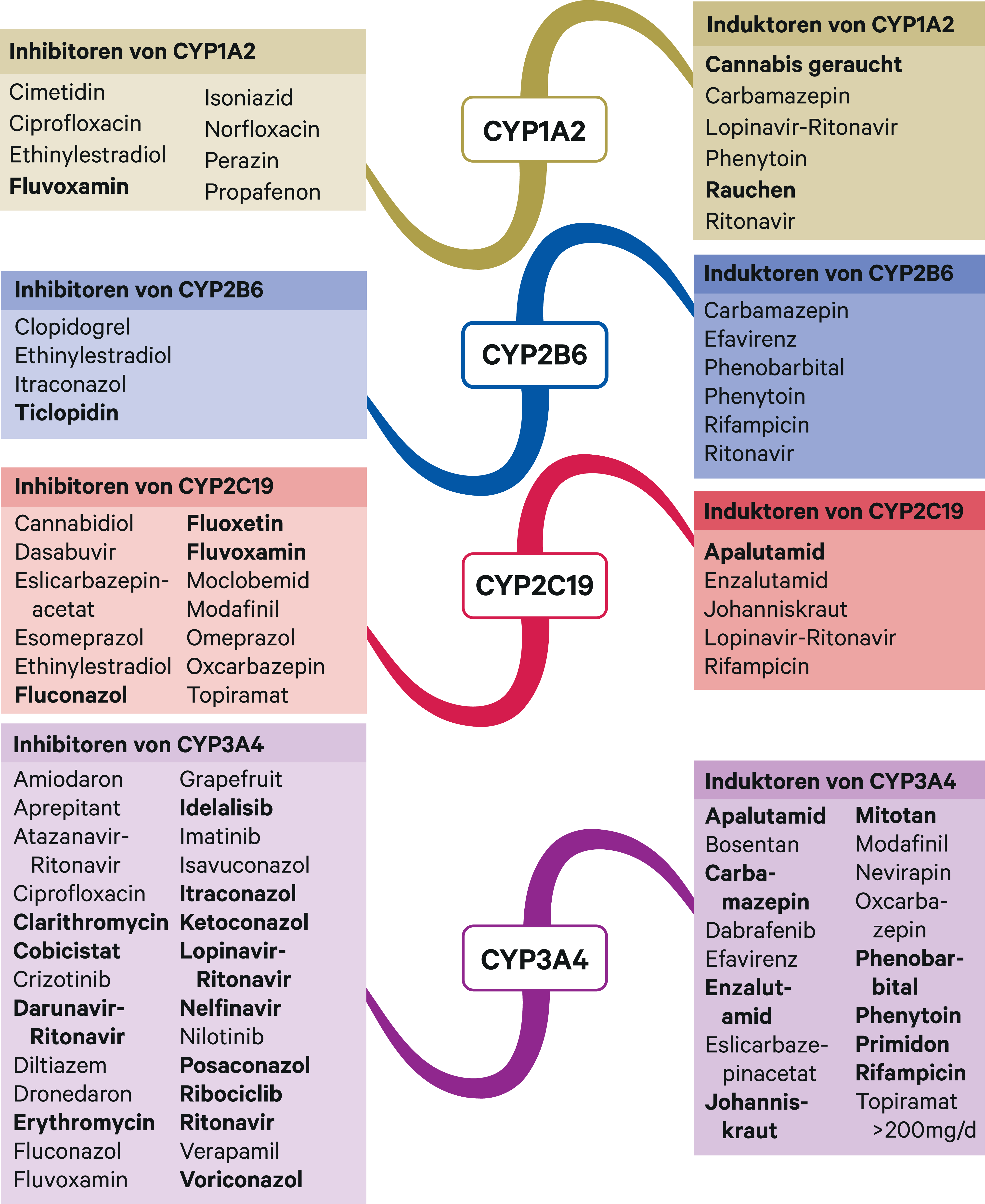
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 1A2, 2B6, 2C19 und 3A4 (Stand 04/2020) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Ancrenaz V, et al. Pharmacokinetic interaction between prasugrel and ritonavir in healthy volunteers. Basic Clin Pharmacol Toxicol 2013;112:132–7.
2. Dalvie DK, O’Connell TN. Characterization of novel dihydrothienopyridinium and thienopyridinium metabolites of ticlopidine in vitro: role of peroxidases, cytochromes p450, and monoamine oxidases. Drug Metab Dispos 2004;32:49–57.
3. Fachinformation Aspirin® N 100 mg. Stand: März 2017.
4. Fachinformation Brilique®. Stand: September 2019.
5. Fachinformation Efient®. Stand: Dezember 2019.
6. Fachinformation Kengrexal®. Stand: September 2016.
7. Fachinformation Plavix®. Stand: November 2019.
8. Fachinformation Tiklyd®. Stand: Oktober 2016.
9. Farid NA, et al. Effect of rifampin on the pharmacokinetics and pharmacodynamics of prasugrel in healthy male subjects. Curr Med Res Opin 2009;25:1821–9.
10. Farid NA, et al. Cytochrome P450 3A inhibition by ketoconazole affects prasugrel and clopidogrel pharmacokinetics and pharmacodynamics differently. Clin Pharmacol Ther 2007;81:735–41.
11. García-Lagunar MH, et al. Genotyping of six clopidogrel-metabolizing enzyme polymorphisms has a minor role in the assessment of platelet reactivity in patients with acute coronary syndrome. Anatol J Cardiol 2017;17:303–12.
12. Gebhard DF. Genetische Determinanten der Clopidogrel-Response-Untersuchungen zu CYP2C19*2 und CYP2C19*17. Dissertation Technische Universität München 2017.
13. Geisslinger G, et al. Mutschler Arzneimittelwirkungen, 11 Aufl. Stuttgart: Wissenschaftliche Verlagsgesellschaft Stuttgart, 2019.
14. Gurbel PA, et al. Clopidogrel efficacy and cigarette smoking status. JAMA 2012;307:2495–6.
15. Itkonen MK, et al. Clopidogrel increases dasabuvir exposure with or without ritonavir, and ritonavir inhibits the bioactivation of clopidogrel. Clin Pharmacol Ther 2019;105:219–28.
16. Karaźniewicz-Łada M, et al. Impact of genetic variants of selected cytochrome P450 isoenzymes on pharmacokinetics and pharmacodynamics of clopidogrel in patients co-treated with atorvastatin or rosuvastatin. Eur J Clin Pharmacol 2020;76:419–30.
17. Kazui M, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos 2010;38:92–9.
18. Kroon LA, Drug interactions with smoking. AM J Health Syst Pharm 2007;64:1971–21.
19. Marsousi N, et al. Impact of boosted antiretroviral therapy on the pharmacokinetics and efficacy of clopidogrel and prasugrel active metabolites. Clin Pharmacokinet 2018;57:1347–54
20. Schwabe U, et al. Arzneiverordnungs-Report 2019. Heidelberg: Springer Verlag, 2019.
21. Scott SA, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther 2013;94:317–23.
22. Shinde DD, et al. Different effects of clopidogrel and clarithromycin on the enantioselective pharmacokinetics of sibutramine and its active metabolites in healthy subjects. J Clin Pharmacol 2013;53:550–8.
23. Teng R, Butler K. Effect of the CYP3A inhibitors, diltiazem and ketoconazole, on ticagrelor pharmacokinetics in healthy volunteers. J Drug Assess 2013;15:30–9.
24. Teng R, et al. Effect of rifampicin on the pharmacokinetics and pharmacodynamics of ticagrelor in healthy subjects. Eur J Clin Pharmacol 2013;69:877–83.
25. Tornio A, et al. Glucuronidation converts Clopidogrel to a strong time‐dependent inhibitor of CYP2C8: A Phase II Metabolite as a perpetrator of drug-drug interactions. Clin Pharmacol Ther 2014;96:498–507.
26. Zhu Y, Zhou J. Identification of the significant involvement and mechanistic role of CYP3A4/5 in clopidogrel bioactivation. ACS Med Chem Lett 2012;3:844–9.
*Nachdruck aus Krankenhauspharmazie 2020;41:192–6.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2020; 27(03)