Holger Petri, Bad Wildungen*
Diazepam
Das Benzodiazepin Diazepam ist nach der Marktrücknahme von Tetrazepam der einzige Vertreter dieser Stoffgruppe zur Behandlung von Zuständen mit erhöhtem Muskeltonus. Der metabolische Abbau von Diazepam erfolgt über Demethylierung und Hydroxylierung zu den pharmakologisch aktiven Metaboliten N-Desmethyldiazepam (Nordazepam), Temazepam und Oxazepam, die dann als Glucuronide ausgeschieden werden [2]. Hieran beteiligt sind das polymorph exprimierte CYP-Isoenzym 2C19 sowie CYP3A4 (Abb. 1). Die Halbwertszeiten der Metaboliten liegen bei 30 bis 100 Stunden für N-Desmethyldiazepam, 10 bis 20 Stunden für Temazepam und 5 bis 15 Stunden für Oxazepam. Dies macht deutlich, dass pharmakokinetische In-vivo-Studien von Diazepam im Steady-State nicht zuletzt wegen des Abhängigkeitsrisikos nur schwer umsetzbar sind.
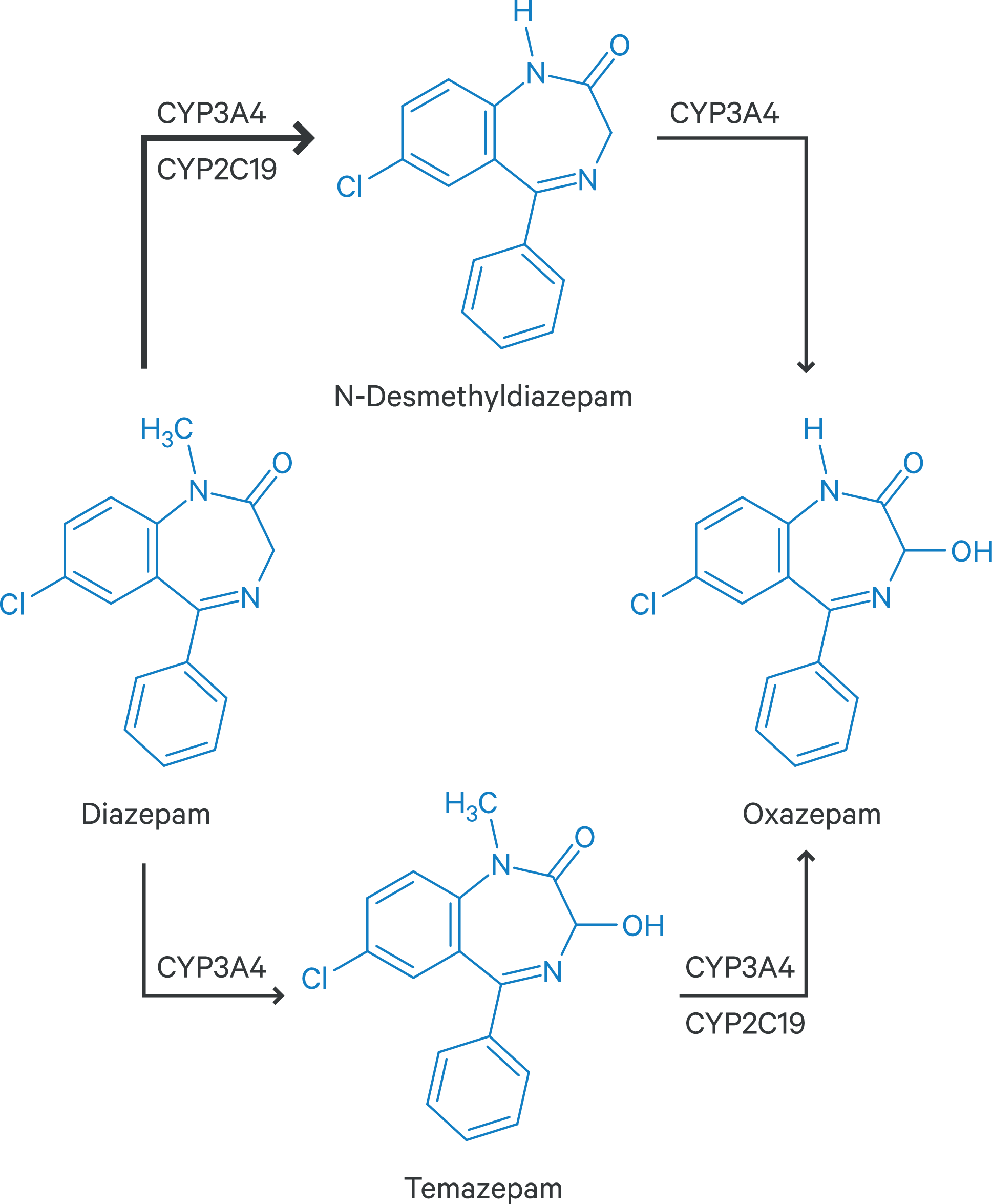
Abb. 1. Phase-I-Metabolismus von Diazepam [19]
In einer klinischen Studie mit gesunden Probanden wurde der Einfluss des CYP3A4-Inhibitors Diltiazem auf die Exposition von Diazepam und des Hauptmetaboliten N-Desmethyldiazepam in Abhängigkeit des CYP2C19-Metabolizer-Status nach Einmalgabe des Benzodiazepins untersucht [8]. Diltiazem steigerte die AUC (Fläche unter der Konzentrations-Zeit-Kurve) von Diazepam und N-Desmethyldiazepam sowohl bei extensiven (EM) als auch langsamen Metabolisierern (PM) in Summe um 21 %. Es ist hervorzuheben, dass der Summenspiegel unter CYP3A4-Inhibition bei gleichzeitigem PM-Status um 71 % höher lag, verglichen mit dem Wert bei Probanden mit EM-Status ohne Einnahme eines CYP3A4-Inhibitors. Unter Steady-State-Bedingungen und der damit verbundenen Kumulation des Hauptmetaboliten (Abb. 1) könnte das Ausmaß in Kombination mit starken CYP3A4-Inhibitoren noch deutlicher ausgeprägt sein.
Bei Kombination von Diazepam und dem starken CYP2C19-Hemmer Fluoxetin (Abb. 2) wurde in neuropsychologischen Tests eine verminderte Aufmerksamkeit festgestellt im Vergleich zur alleinigen Gabe von Diazepam oder Fluoxetin [10]. Bei Kombination von Diazepam (einmalig 10 mg) und Fluvoxamin, einem weiteren starken CYP2C19-Inhibitor, sank die Clearance von Diazepam um 65 % und die Halbwertszeit verdoppelte sich in etwa von 51 auf 118 Stunden. Die Blutspiegel von Diazepam und N-Desmethyldiazapem stiegen. Unter der Kombination ist mit vertiefter und verlängerter Sedierung zu rechnen [13].
In einer weiteren pharmakokinetischen Studie wurde der Einfluss des CYP-Induktors Rifampicin (Abb. 2) auf die Exposition von Diazepam und N-Desmethyldiazepam untersucht. Die Bioverfügbarkeit von Diazepam sank um 77 % und die des Metaboliten um die Hälfte. In Summe reduzierte sich die Exposition um zwei Drittel [11]. Der therapeutische Effekt als Muskelrelaxans kann daher reduziert sein.
Daten zum Einfluss von CYP3A4-Induktoren bei Patienten mit einem ultraschnellen CYP2C19-Metabolisierer-Status liegen nicht vor.
Tizanidin und Orphenadrin
Tizanidin wird primär über CYP1A2 verstoffwechselt. In einer klinischen Studie mit gesunden Probanden erhöhte sich der AUC-Wert von Tizanidin durch den starken CYP1A2-Hemmer Fluvoxamin (Abb. 2) durchschnittlich um das 33-Fache [6]. Ciprofloxacin hat die Tizanidin-Bioverfügbarkeit um das 10-Fache gesteigert [5].
Daraus kann sich eine klinisch bedeutsame, lang anhaltende Blutdrucksenkung mit Schläfrigkeit, Schwindel und, aufgrund der starken anticholinergen Eigenschaften, verminderte psychomotorische Leistungsfähigkeit ergeben. Potente CYP1A2-Inhibitoren wie Fluvoxamin und Ciprofloxacin sind unter einer Tizanidin-Therapie kontraindiziert [4].
Polyzyklische Kohlenwasserstoffe, die durch die Verbrennungsprozesse beim Rauchen gebildet werden, induzieren die Bildung von CYP1A2 [20]. In einer Studie lagen die AUC-Werte von Tizanidin bei rauchenden Probanden um 33 % niedriger im Vergleich zu Nichtrauchern [1]. Der Abfall der Plasmaspiegel fällt moderat aus, kann aber im Einzelfall eine Dosiserhöhung notwendig machen; besonders in der Langzeittherapie bei männlichen Rauchern [4].
Oxycodon wird über CYP2D6 zu Oxymorphon abgebaut. In einer umfassenden In-vitro-Untersuchung hatten Tizanidin und Orphenadrin eine niedrige Hemmkonzentration (Ki) in Bezug auf CYP2D6 und blockierten damit den Metabolismus des Opioids [9]. Orphenadrin zeigte schon in einer älteren In-vitro-Studie eine starke CYP2D6-Hemmung [7]. Die beiden Prodrugs Tamoxifen und Tramadol, die erst über CYP2D6 zu den pharmakologisch aktiven Metaboliten verstoffwechselt werden, können in ihrer Wirksamkeit klinisch relevant beeinträchtigt sein [14, 15].
Die Datenlage zum Metabolismus von Orphenadrin ist spärlich. CYP-Enzyme sind am Metabolismus beteiligt [16], die Bedeutung pharmakokinetischer Wechselwirkungen ist aber nicht bekannt.
Tolperison
Der Abbau von Tolperison erfolgt über das polymorph exprimierte CYP2D6 [12]. Eine Dosisreduktion nach klinischem Ansprechen kann bei gleichzeitiger Therapie mit CYP2D6-Hemmern (Abb. 2) erwogen werden. Gemäß Herstellerangaben sind Arzneimittelinteraktionen, nicht zuletzt wegen der therapeutischen Breite, bisher nicht beobachtet worden. Über eine kompetitive Hemmung sind Wechselwirkungen mit anderen Substraten, die primär über CYP2D6 metabolisiert werden, nicht auszuschließen [3].
Baclofen, Methocarbamol und Pridinol
Diese zentralen Muskelrelaxanzien werden teils unverändert und teils über Phase-II-Reaktionsschritte metabolisiert ausgeschieden [17, 18].
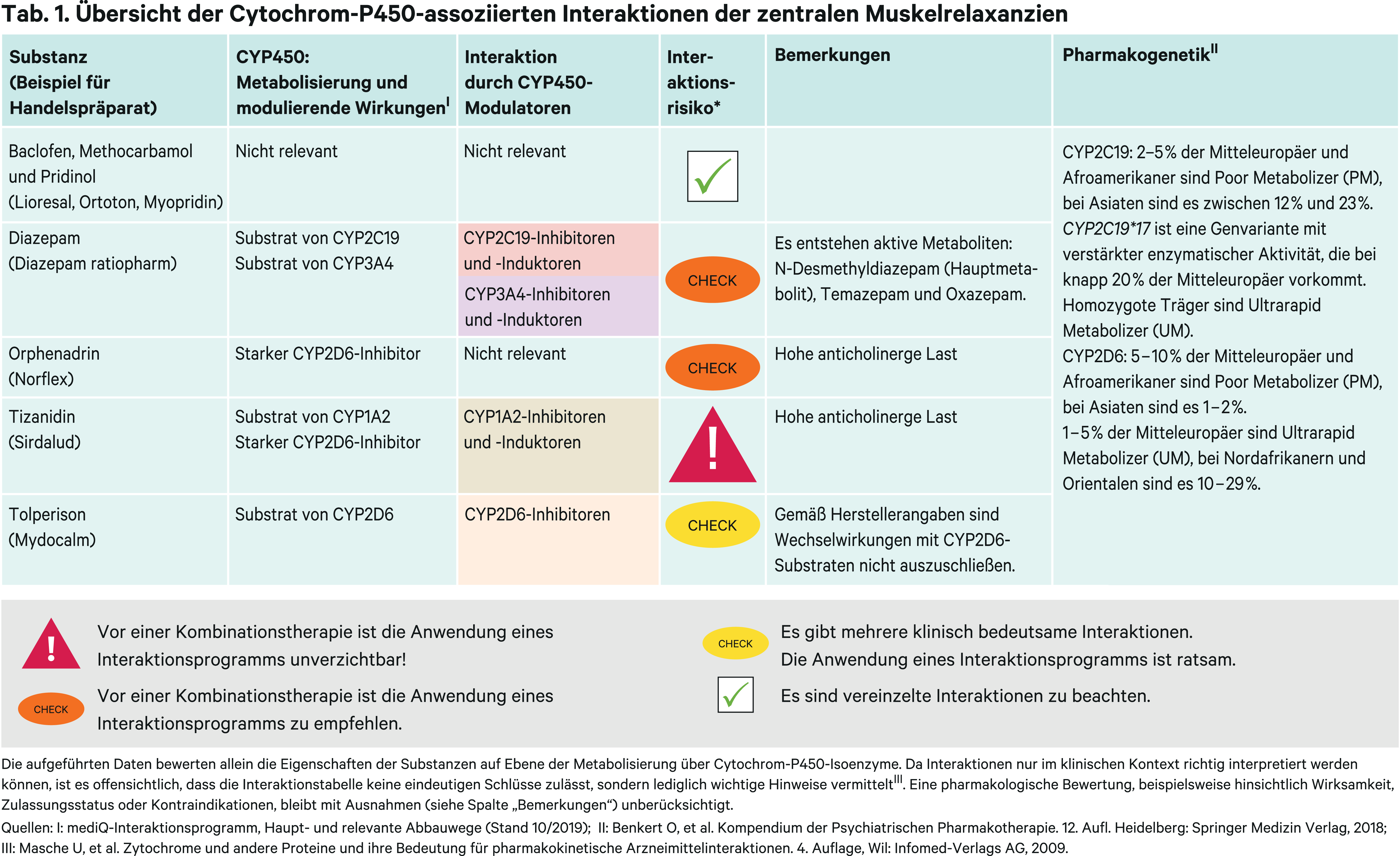
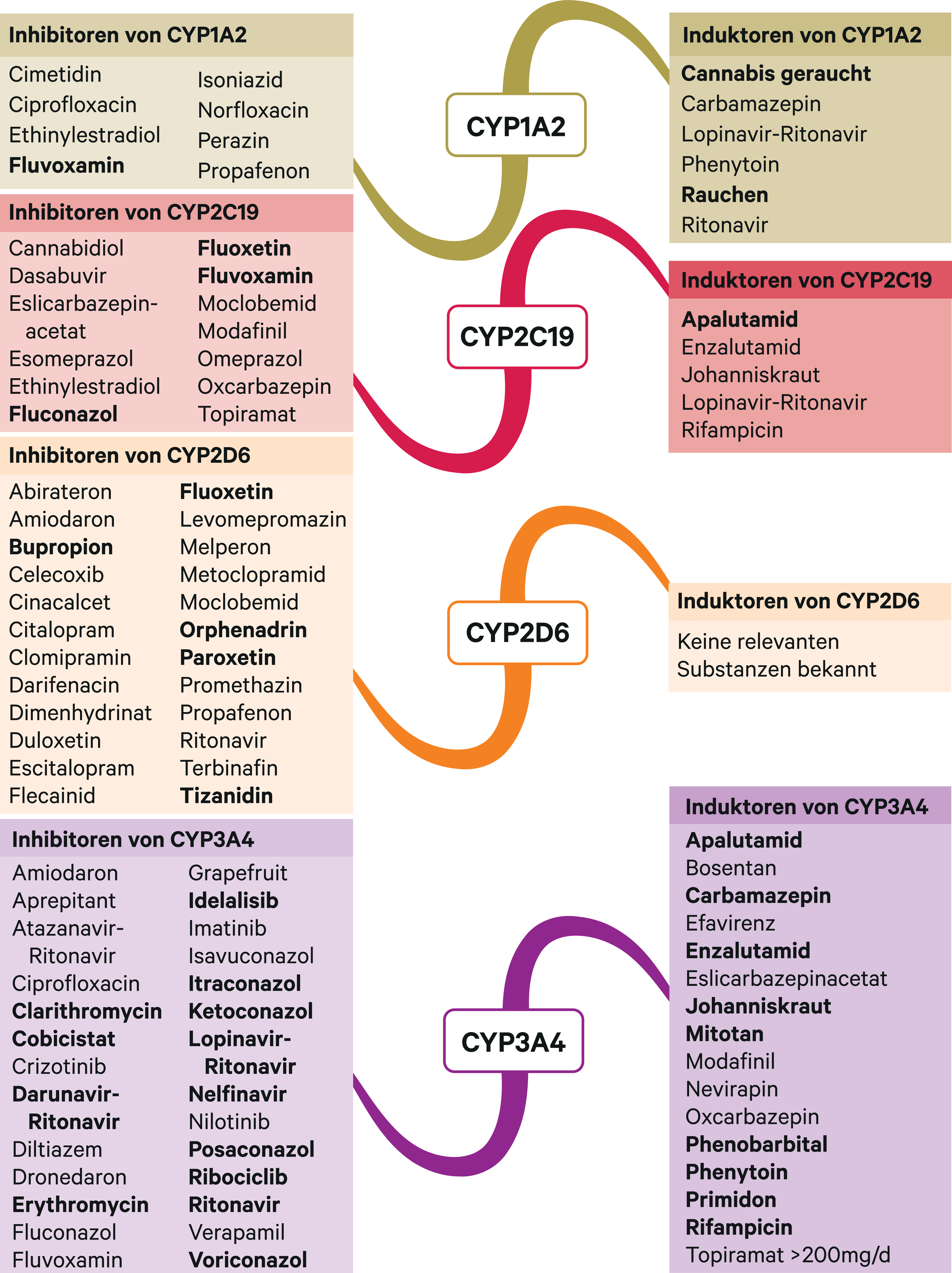
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 1A2, 2C19, 2D6 und 3A4 (Stand 10/2019) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Backman JT, et al. Effects of gender and moderate smoking on the pharmacokinetics and effects of the CYP1A2 substrate tizanidine. Eur J Clin Pharmacol 2008;1:17–24.
2. Fachinformation Diazepam ratiopharm®. Stand: April 2019.
3. Fachinformation Mydocalm®. Stand: Januar 2019.
4. Fachinformation Sirdalud®. Stand: Februar 2019.
5. Granfors MT, et al. Ciprofloxacin greatly increases concentrations and hypotensive effect of tizanidine by inhibiting its cytochrome P4501A2-mediated presystemic metabolism. Clin Pharmacol Ther 2004;6:598–606.
6. Granfors MT, et al. Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction. Clin Pharmacol Ther 2004;4:331–41.
7. Guo Z, et al. Orphenadrine and methimazole inhibit multiple cytochrome P450 enzymes in human liver microsomes. Drug Metab Dispos 1997 Mar;25:390–3.
8. Kosuge K, et al. Effects of CYP3A4 inhibition by diltiazem on pharmacokinetics and dynamics of diazepam in relation to CYP2C19 genotype status. Drug Metab Dispos 2001;10:1284–9.
9. Moody DE, et al. Inhibition of in vitro metabolism of opioids by skeletal muscle relaxants. Basic Clin Pharmacol Toxicol 2018;123:327–34.
10. Moskowitz H, Burns M. The effects on performance of two antidepressants, alone and in combination with diazepam. Prog Neuropsychopharmacol Biol Psychiatry 1988;12:783–92.
11. Ohnhaus EE, et al. The effect of antipyrine and rifampin on the metabolism of diazepam. Clin Pharmacol Ther 1987;42:148-56.
12. Pawlowska M, et al. Influence of CYP2D6 and CYP2C19 genetic polymorphism on the pharmacokinetics of tolperisone in healthy volunteers. Eur J Clin Pharmacol 2015;6:699–705.
13. Perucca E, et al. Inhibition of diazepam metabolism by fluvoxamine: a pharmacokinetic study in normal volunteers. Clin Pharmacol Ther 1994;56:471–6.
14. Petri H, Grandt D. Interaktionen der Opioidanalgetika auf Ebene der Biotransformation. Schmerz 2016;30:519–25.
15. Petri H. Das Interaktionspotenzial der Antiestrogene. Krankenhauspharmazie 2017;38:152–5.
16. Roos PH, Mahnke A. Metabolite complex formation of orphenadrine with cytochrome P450. Involvement of CYP2C11 and CYP3A isozymes. Biochem Pharmacol 1996;52:73–84.
17. www.fachinfo.de (letzter Zugriff am 26.09.2019).
18. www.mediq.ch (letzter Zugriff am 26.09.2019).
19. www.pharmgkb.org/pathway/PA165111375 (letzter Zugriff am 16.10.2019).
20. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet 1999;36:425–38.
*Nachdruck aus Krankenhauspharmazie 2019;40:520–3.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2019; 26(06)