Hans-Jürgen Möller, München, und Karl Broich, Bonn
Teil I (Allgemeine Grundlagen, 4-Phasen-Modell, Design klinischer Prüfungen) ist in Heft 5/2018 erschienen (Psychopharmakotherapie 2018;25:251–7)
Selektion der Stichprobe
In Studien einbezogene Patienten stellen immer eine Selektion aus der psychiatrisch zu versorgenden Patientenpopulation sowie eine Selektion der Patienten mit einer bestimmten psychiatrischen Diagnose dar [13]. Dabei hängt die Selektion unter anderem davon ab, ob man beispielsweise für eine Phase-III-Studie rekrutiert oder für eine Phase-IV-Studie. In Phase-III-Studien müssen Patienten eingeschlossen werden, die hochselektiert sind: Altersspanne 18 bis 65 Jahre (bei Studien an erwachsenen Patienten), ausreichender Schweregrad der psychopathologischen Symptomatik (z. B. HAMD-17-Gesamtscore über 22), keine psychische oder somatische Komorbidität, keine Komedikation (abgesehen gegebenenfalls z. B. von einer beschränkten Gabe von Hypnotika/Sedativa), keine Vorgeschichte von Therapieresistenz, keine Chronizität der Symptomatik und ähnliches. Diese Ausschlusskriterien dienen dazu, in einer solchen experimentellen Studie Störvariablen weitestgehend auszuschließen und die „interne Validität“ der Studie möglichst optimal zu garantieren. In einer Phase-IV-Studie hingegen fallen die meisten dieser Ausschlusskriterien weg, da man eine möglichst der üblichen Versorgungssituation entsprechende Stichprobe rekrutieren will und deshalb alle komplizierenden Faktoren/Einflüsse akzeptiert. Es geht in entsprechend geplanten Phase-IV-Studien um die Generalisierbarkeit der Ergebnisse (externe Validität), nicht mehr um den prinzipiellen Wirksamkeitsnachweis unter abstrahierenden experimentellen Bedingungen.
Die richtige Selektion der Patienten für eine klinische Prüfung ist von besonderer Wichtigkeit. Eine nicht dem Prüfziel entsprechende Auswahl der Patienten kann von vornherein zu einem Misserfolg der klinischen Studie führen (s. u.!). Depressive Patienten beispielsweise, die klinisch stationär behandelt werden, entsprechen hinsichtlich der Ansprechbarkeit auf Antidepressiva in der Regel nicht den ambulant behandelten Patienten, da Patienten, die schlecht auf Antidepressiva ansprechen, in der Klinik überrepräsentiert sind. Fasst man die „Antidepressiva-Nonresponder“ als eine spezielle Subpopulation der Depressiven auf, so muss man in Erwägung ziehen, dass in der Klinik antidepressive Substanzen an Personen geprüft werden könnten, denen nicht ausreichend mit Antidepressiva geholfen werden kann. Das führt eventuell dazu, dass die Prüfsubstanz keine Überlegenheit zeigen kann. Eine solche Selektion von schlecht auf Antidepressiva ansprechenden Patienten wäre für die Psychopharmaka-Prüfung nur die richtige Stichprobe, wenn man ein Antidepressivum oder eine Kombinationsbehandlung (z. B. Antidepressivum plus Antipsychotikum) in dieser speziellen Indikation prüfen wollte.
Zunehmend wurde wegen der dargestellten Problematik und vor allem auch aus Praktikabilitätsgründen (schnellere Rekrutierungsmöglichkeit) die Prüfung der meisten neuen Antidepressiva und anderen Psychopharmaka vorrangig an ambulanten Patienten durchgeführt. Allerdings hat das offensichtlich zu neuen Problemen geführt, beispielsweise einem vermehrten Ansprechen auf Placebo und damit gegebenenfalls einer geringeren Placebo-Verum-Differenz bei Antidepressiva-Studien, möglicherweise unter anderem dadurch bedingt, dass mehr Patienten mit Symptomatik geringeren Intensitätsgrads eingeschlossen werden [16]. Um diese Problematik zu umgehen, werden heutzutage für Antidepressiva-Studien bzw. bei Prüfung anderer Psychopharmaka gezielt Patienten mit einem ausreichendem Schweregrad der Depression oder sonstigen Symptomatik rekrutiert.
Neben solchen Aspekten der Patientenauswahl im Rahmen von Studien ist zu berücksichtigen: Je stärker ein Untersuchungsansatz standardisiert ist und je einschränkender er in den therapeutischen Möglichkeiten ist, desto enger ist auch die Patientenselektion. Diesbezüglich gehen beispielsweise Placebo-kontrollierte Studien mit der stärksten Patientenselektion einher, das heißt, viele Patienten (u. a. Patienten mit einem zu hohem Schweregrad der Depression, Patienten mit deutlicher Suizidalität) müssen von solchen Studien ausgeschlossen werden, während Prüfungen gegen ein Standardpräparat noch durchführbar wären. Einfache Anwendungsbeobachtungen („non-interventional studies“, NIS) sind weitgehend voraussetzungsfrei für die einzuschließenden Patienten. Hinsichtlich der Patienten-Selektion stellen die „Real-World“-Studien einen methodischen Mittelweg zwischen Phase-III-Studien und einfachen Anwendungsbeobachtungen dar [15]. Wie schon in Teil I dieser Publikation betont [14], hat jeder dieser unterschiedlichen Studien-Ansätze eine unterschiedliche Zielsetzung und ein unterschiedliches Bias-/Fehler-Risiko. In der Regel können Ergebnisse aus Studien mit einem größeren Fehler-Risiko bzw. einer geringeren internen Validität Ergebnisse von Studien mit geringerem Fehler-Risiko nicht falsifizieren, sondern nur komplementäre Daten liefern. Das gilt insbesondere für das Verhältnis von Phase-IV- zu Phase-III-Ergebnissen [15].
Hypothesenprüfung, Studien-Endpunkte, Signifikanz, „Power“-Kalkulation
Phase-III-Prüfungen sind immer Hypothesen prüfend, und zwar in der Regel hinsichtlich der Prüfung der Überlegenheit. Daneben gibt es auch die Prüfung auf Gleichheit (siehe Teil 1 dieser Darstellung [14]), die aber unter vielen Aspekten problematisch ist und deshalb hier nicht weiter hinsichtlich der Statistik dargestellt wird. Es gibt auch rein deskriptive Untersuchungen ohne Hypothesenformulierung, beispielsweise viele Phase-IV-Studienansätze (u. a. Erfassung von Nebenwirkungen im alltäglichen Gebrauch eines Medikaments). Im Folgenden wird die Prüfung auf Wirksamkeit (und zwar in der Akutbehandlung) exemplarisch in den Vordergrund gestellt, Sicherheits- und Verträglichkeitsaspekte werden aus Platzgründen nicht weiter berücksichtigt. Auch auf Besonderheiten der Wirksamkeits- und Verträglichkeitsprüfung in Langzeitstudien, beispielsweise Studien zur Erhaltungstherapie/Rezidivprophylaxe, kann aus Platzgründen nicht weiter eingegangen werden.
Bei Hypothesen prüfenden Untersuchungen wird vor Beginn der Prüfung (a priori) eine Hypothese formuliert, beispielsweise statistisch signifikante Überlegenheit des Antidepressivums A gegen Placebo im Mittelwertsvergleich des Hauptzielparameters (z. B. HAMD-17-Gesamtscore). Als Signifikanzschwelle wird je nach sonstigen Aspekten, beispielsweise Fallzahl, das 0,05- (5 %) oder 0,01- (1 %) Niveau festgelegt. Gegebenenfalls gibt es noch einen weiteren primären Hauptzielparameter, für den auch eine Hypothese formuliert wird. Es besteht auch die Möglichkeit, weitere Hypothesen nach der Formulierung des Prüfplans bzw. nach Fertigstellung der Studie („ex post“, s. u.) zu formulieren, deren statistische Prüfung dann aber nur heuristischen Wert hat. Ihre konfirmatorische Prüfung bedarf einer eigenen neuen Untersuchung mit A-priori-Formulierung dieser Hypothese. Diese präzisere Form experimentellen Denkens wird leider im Kontext kaum noch überschaubarer metaanalytischer Auswertungen großer Datensätze von zahleichen Studien nicht beachtet, bei denen immer wieder neue Details als Ergebnisse dargestellt werden, ohne den experimentellen Prüfzusammenhang zu berücksichtigen.
Als Studienendpunkte bezeichnet man Parameter, die nach Abschluss der Studie für die statistische Auswertung vorgesehen sind. Sie werden a priori festgelegt, auch ob sie primäre oder sekundäre Endpunkte sind. Primäre Endpunkte sind die, auf die sich die Haupthypothese bezieht (z. B. Mittelwertsunterschied in einer Depressionsskala), in der Regel einer oder zwei (z. B. als 2. Endpunkt Unterschied in der Responder-Häufigkeit). Sekundäre Endpunkte können zahlreicher sein, beispielsweise Clinical Global Impression (CGI) oder ähnliche Skalen, Selbstbeurteilungsskalen, Skalen zur sozialen Adaptation oder zur Lebensqualität. Wird nach Abschluss des Prüfplans/der Studie beschlossen, dass weitere Parameter, deren statistische Analyse ursprünglich nicht geplant war, ausgewertet werden sollen, so spricht man von einem Ex-post-Ansatz. Diese Differenzierungen sind sehr wichtig, wie gleich im Zusammenhang mit der Fallzahlschätzung gezeigt wird.
Zunächst noch die Erklärung zweier statistischer Grundbegriffe, die in diesem Zusammenhang wichtig sind. Als Alpha-Fehler (Fehler erster Art) bezeichnet man in der Statistik das Verwerfen der Nullhypothese (= die Annahme, dass zwischen Experimentalgruppe und Kontrollgruppe kein Unterschied besteht), obwohl sie in Wirklichkeit richtig ist. Die Wahrscheinlichkeit, einen Alpha-Fehler zu machen, ist kleiner oder gleich dem Signifikanzniveau Alpha. Als Beta-Fehler (Fehler zweiter Art) bezeichnet man in der Statistik, dass zu Unrecht die Nullhypothese angenommen wird.
Die Bedeutung eines statistisch signifikanten Unterschieds in einem primären Wirksamkeitsparameter ist wesentlich höher einzuschätzen als jene in einem oder mehreren anderen sekundären Wirksamkeitsparametern. Das gilt noch mehr für weitere ex post definierte Endpunkte. Das hängt mit den Prinzipien der Fallzahlschätzung zusammen (Beispiel 1).
Beispiel 1
In einer Studie Antidepressivum vs. Placebo soll ein Mittelwerts-Unterschied von 3 Punkten auf der Montgomery-Asberg Depression Rating-Skala (MADRS) auf einem Signifikanzniveau von 5 % (≤ 0,05; Fehler erster Art), einem Beta-Fehler von 10 %, einer Power von 90 % (s. u.), bei einer angenommenen Streuung von 5 Punkten der MADRS zum Studienendpunkt entdeckt werden. Eingesetzt in eine entsprechende Formel zur Fallzahlberechnung resultiert dann eine Gruppengröße von etwa 300 Patienten pro Gruppe. Das gegebenenfalls positive Ergebnis ist statistisch beweisend, aber natürlich immer noch mit einer Irrtumswahrscheinlichkeit von 5 % behaftet.
Als Power bezeichnet man die Wahrscheinlichkeit, mit der ein Unterschied in der gegebenen Prüfung gefunden wird, wenn er existiert. Je größer die Power, je kleiner der Alpha-Wert und je größer der als klinisch relevant angenommene Unterschied veranschlagt werden, desto größer ist der notwendige Stichprobenumfang. Für sekundäre Wirksamkeitsparameter ist diese Beweiskraft wesentlich geringer, da die Gruppengröße nicht darauf berechnet wurde. Das gilt noch mehr für weitere ex post festgelegte Endpunkte. Obendrein erhöht sich die Wahrscheinlichkeit, dass ein signifikantes Ergebnis fälschlicherweise gefunden wird, mit jeder weiter durchgeführten Testung. Das wird als Problem des multiplen Testens bezeichnet und macht die sogenannte Alpha-Adjustierung erforderlich. Um das Ergebnis einer Studie zu sichern, ist eine zweite gleichangelegte Studie erforderlich. Wenn auch diese positiv ausfällt, so kann mit hoher Wahrscheinlichkeit die Wirksamkeit angenommen werden.
Statistische Auswertung, OC- und ITT-(LOCF-)Analysen, statistische Signifikanz, Effektstärke
Je nach Art der Studien kommen bei der Auswertung unterschiedliche statistische Verfahren zur Anwendung, bei denen in Akutstudien der Mittelwertsvergleich von Skalenwerten bzw. der Häufigkeitsvergleich von Respondern und in Langzeitstudien der Häufigkeitsvergleich von Rückfällen im Zentrum steht. In Langzeitstudien zur Erhaltungstherapie/Rezidivprophylaxe wird gern auch die Survival-Analyse (Abb. 5–6) angewendet, die darstellt, wie lange die Patienten der jeweiligen Gruppe ohne Rückfall waren [13, 21]. Jeder statistische Signifikanz-Test beruht auf Voraussetzungen, die zunächst geprüft werden müssen. Werden diese verletzt, so ist der Test in der Regel nicht anwendbar. Bei allen statistischen Auswertungsmethoden, die auf Mittelwertvergleichen von Stichproben beruhen, muss damit gerechnet werden, dass man durch die Reduktion der Daten auf Mittelwerte erhebliche Informationsverluste hinnimmt. Durch zusätzliche statistische Analysen sollte versucht werden, derartige durch Mittelwertbildung bedingte Informationsverluste zu kompensieren.
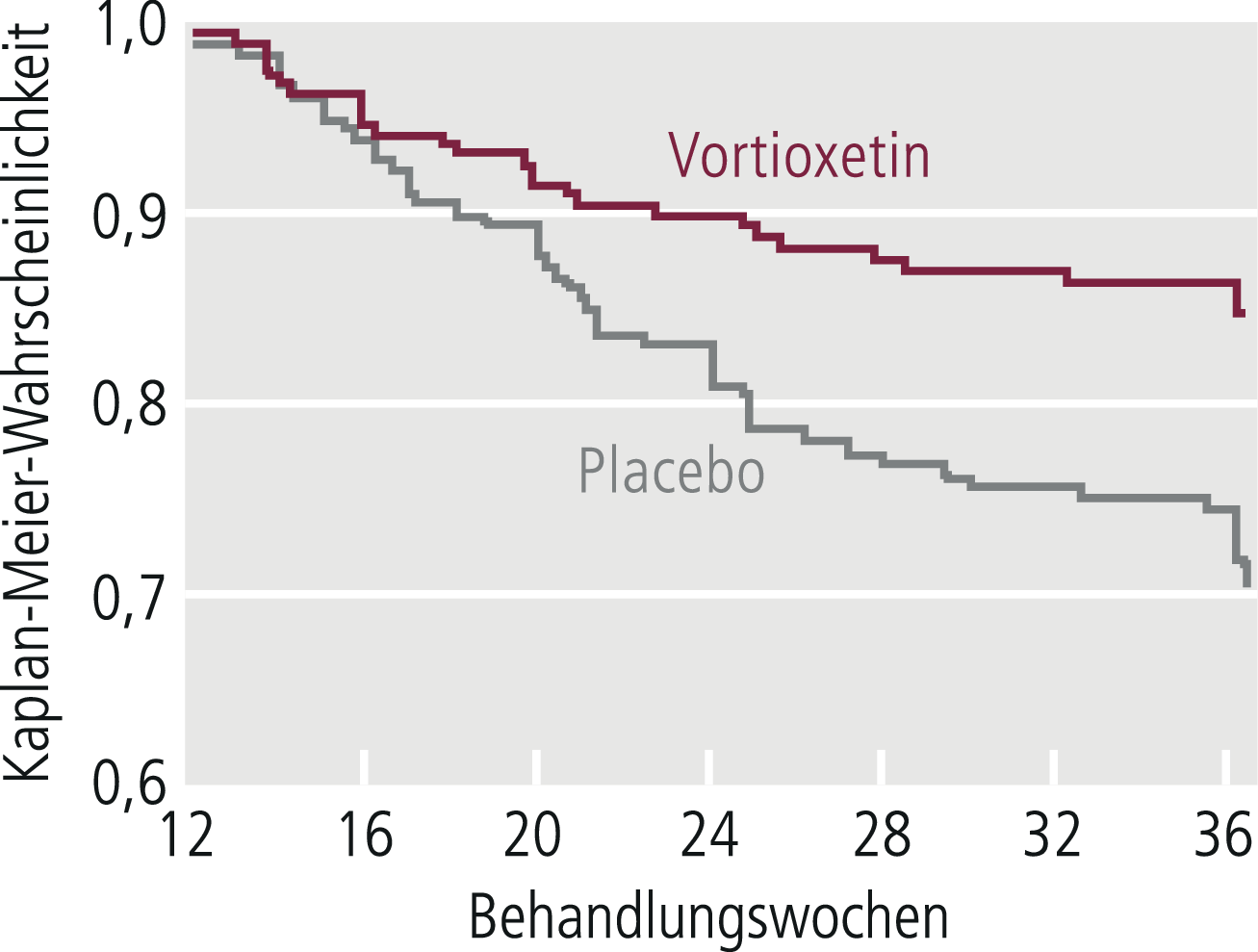
Abb. 5–6. Kaplan-Meier-Survival-Analyse während der doppelblinden Langzeitstudie an Remittern aus einer Akutstudie zu Vortioxetin bei Major Depressive Disorder (mod. nach [2])
Wichtig ist, dass neben der „Observed-Case-Analyse“ (OC-Analyse), die sich auf die Patienten bezieht, die die Therapiephase absolviert haben, auch die „Intent-to-treat-Analyse“ (ITT-Analyse) durchgeführt wird, die auch die „Drop-outs“ (= Studienabbrecher) einbezieht und somit alle Patienten berücksichtigt [21]. Die OC-Analyse geht mit einer Reduktion der Studienstichprobe einher. Sie gibt Auskunft darüber, wie gut das Ansprechen prinzipiell möglich ist, wenn ein Patient durchgehend die Medikation eingenommen hat. Diese Methode kann zur Überschätzung der Wirksamkeit im Vergleich zur ITT-Analyse führen. Die in ihren Ergebnissen im Vergleich zur OC-Analyse weniger optimistische ITT-Analyse wird von den Zulassungsbehörden als Hauptentscheidungskriterium herangezogen, sofern diese als konservativ angenommen werden können (Abb. 5–7). Bei der ITT-Analyse wird von den Patienten, die vorzeitig ausgeschieden sind, der jeweils letzte beobachtete Wert des zu untersuchenden Zielparameters von Patienten weitergeführt und geht so in die Auswertung der Daten mit ein („last observation carried forward“, LOCF). In den letzten Jahren wurde zunehmend anstelle der LOCF-Methode die „Mixed effect model repeated measures“(MMRM)-Methode vorgeschlagen, die unter einigen Aspekten sinnvoller scheint [12]. In gewisser Weise stellt der MMRM-Ansatz einen Mittelweg zwischen der OC-Analyse und der ITT-LOCF-Analyse dar.
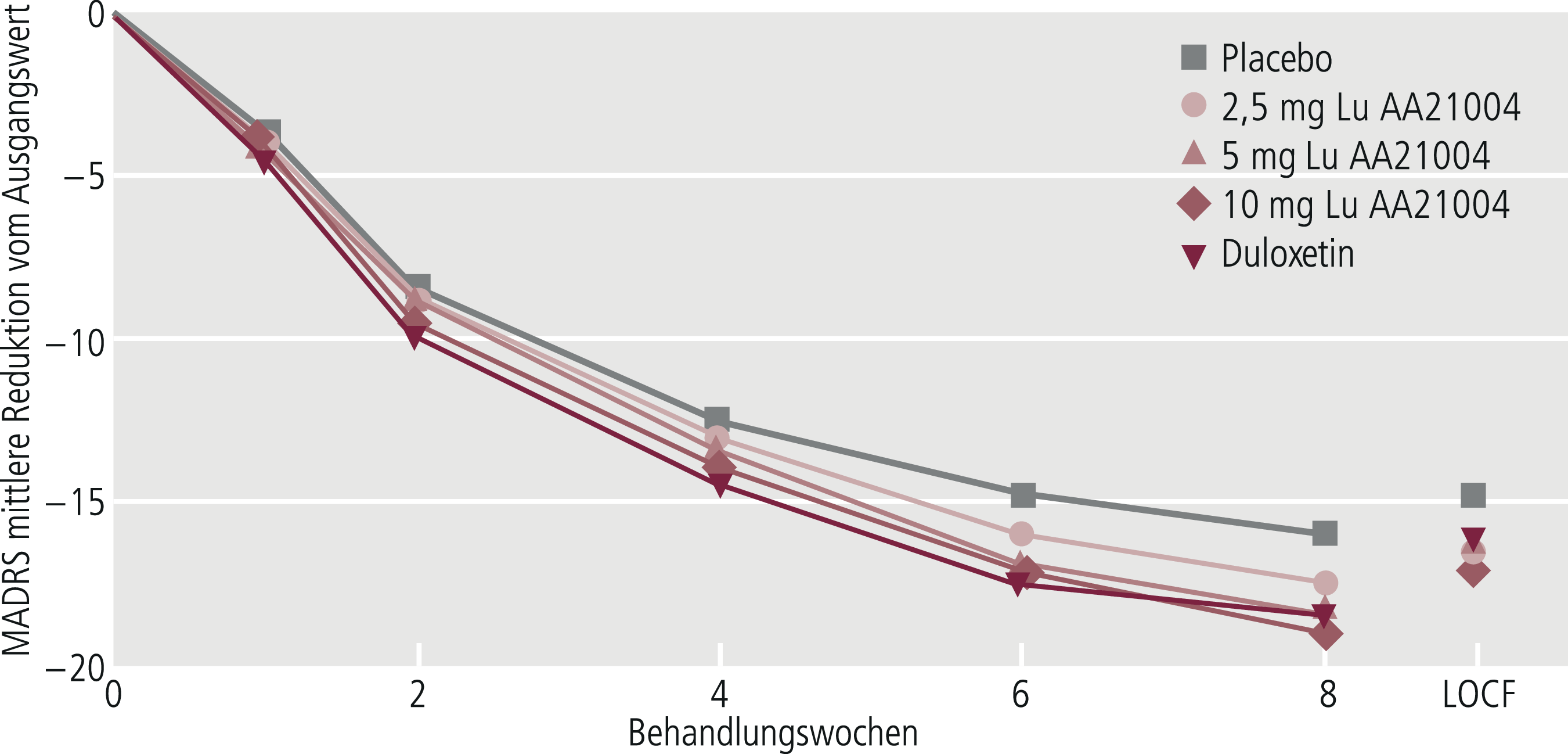
Abb. 5–7. Vergleich des Observed-Case-(OC-)Ansatzes bei Woche 8 mit dem LOCF-Ansatz. Der OC-Ansatz zeigt niedrigere Endwerte und partiell größere Unterschiede zwischen den Gruppen als der LOCF-Ansatz (mod. nach [1])
Kommen verschiedene klinische Studien zu gleichlautenden, statistisch gesicherten Ergebnissen, kann das Ergebnis als gesichert angesehen werden. Zur Beschreibung des Ausmaßes der statistischen Signifikanz werden p-Werte angegeben, beispielsweise 5 % (p ≤ 0,05) oder 1 % (p ≤ 0,01). Zunehmend häufig werden zusätzlich Konfidenzintervalle dargestellt. Zur Kennzeichnung der klinischen Relevanz der Wirksamkeit dient die „Number needed to treat“ (NNT), unterteilt nach „Number needed to treat to benefit“ (NNTB) und „Number needed to harm“ (NNTH, auch NNH). Je geringer der Unterschied zwischen zwei Behandlungsmethoden ist, desto größer ist die NNTB. Zur weiteren Kennzeichnung des Unterschieds der Ergebnisse aus Verum-Gruppe vs. Kontrollgruppe können Effektstärkenmaße, wie sie insbesondere bei Metaanalysen verwendet werden, angegeben werden (Tab. 5–4).
Tab. 5–4. Termini zur Bezeichnung des Ausmaßes der Überlegenheit/Unterlegenheit eines Therapieverfahrens im Vergleich zu einem anderen
|
Statistische Signifikanz: meist als 1%- oder 5%-Niveau ausgedrückt |
|
Konfidenzintervall: Der Bereich, innerhalb dessen ein wahrer Wert (beispielsweise die Effektstärke) bei einer Studienpopulation mit einer gewissen Wahrscheinlichkeit (z. B. 95 % oder 99 %) liegt. Konfidenzintervalle geben die Wahrscheinlichkeit von Zufallsfehlern, nicht jedoch von systematischen Fehlern in Studien wieder. |
|
Number needed to treat (NNT): Anzahl der Menschen, die behandelt werden müssen, um ein Ergebnis (z. B. Responder) zu erzielen. Man kann weiter unterteilen in NNTB (= Number needed to benefit) und NNTH (= Number needed to harm). Je höher die NNT, desto kleiner der Unterschied zwischen zwei Behandlungsverfahren. Wird als Kehrwert des absoluten Risikounterschieds berechnet. NNT = 1/0,01 = 10 |
|
Absoluter Risikounterschied (AR): eine Effektstärke für dichotome Variablen, bei der das Risiko für ein Outcome in der einen Gruppe vom Risiko in der anderen Gruppe abgezogen wird. |
|
Relatives Risiko (RR): eine Effektstärke für dichotome Variablen, bei der das Risiko für einen Outcome in der einen Gruppe vom Risiko in der anderen Gruppe geteilt wird: RR = Risiko Gruppe 1/Risiko Gruppe 2 |
|
Odds-Ratio (OR): Effektstärke für dichotome Variablen. Der Begriff Odds drückt aus, wie häufig das Ereignis in einer Gruppe aufgetreten ist (a) geteilt durch die Häufigkeit des Nichtauftretens in der gleichen Gruppe (b). Das Odds-Ratio vermittelt das Verhältnis von zwei Odds. Ein Wert von 1 besagt, dass die Häufigkeiten von zwei Merkmalen sich nicht unterscheiden. Werte unter und über 1 indizieren eine Assoziation zwischen den beiden Merkmalen. |
|
Gewichteter mittlerer Unterschied („weighted mean difference“ – WMD): Effektstärke für kontinuierliche Variablen. Ein bei Metaanalysen angewendetes Differenzmaß, zu dessen Errechnung verschiedene Messergebnisse aus unterschiedlichen Studien mit bekanntem Mittelwert, Standardabweichungen und Stichprobengröße ermittelt und nach deren Einfluss gewichtet werden. |
|
Standardisierter mittlerer Unterschied („standardised mean difference“ – SMD): Landläufig in Metaanalysen als „Effektstärke“ („effect size“) bezeichnet. Modifikation der Formel für den gewichteten mittleren Unterschied, indem zusätzlich durch die gepoolte Standardabweichung beider Gruppen geteilt wird. Durch das Teilen durch die Standardabweichung eine Standardisierung der Effektstärke: sie wird in ein Maß in Standardabweichungseinheiten verwandelt. Eine alte Faustregel (kritisch zu sehen!) nach Cohen [7] graduiert eine Effektstärke von 0,20 als kleinen, von 0,50 als mittleren und von größer als 0,80 als großen Unterschied zwischen zwei Interventionen. |
Je einheitlicher die Stichprobe bezüglich Diagnose, Erkrankungsdauer, Lebensalter, Ausprägungsgrad der Symptome u. a., desto einfacher ist die Auswertung und desto größer die Wahrscheinlichkeit eindeutiger Ergebnisse. Mit diesen Vorteilen einer homogenen Stichprobe erkauft man sich aber gleichzeitig eine schlechte Übertragbarkeit der Ergebnisse in die therapeutische Praxis aufgrund der mangelnden Repräsentativität für die Grundgesamtheit der zu behandelnden Patienten (fehlende externe Validität). Auch unter dem Aspekt von Erkundungsstudien, die die Interferenz des Pharmakons mit bestimmten Persönlichkeits- oder Krankheitsmerkmalen analysieren, ist die Forderung nach Homogenität der Stichprobe einzuschränken. Hier kann gerade eine sehr heterogene Stichprobe intensiver zur Hypothesengeneration anregen als eine bezüglich der Merkmalspluralität reduzierte.
Analyse der Wirkfaktoren
In der klinischen psychopharmakologischen Forschung wie überhaupt in der Therapieforschung bei psychisch Kranken besteht die schwierige Situation, dass die unabhängige Variable, die experimentell variiert bzw. manipuliert wird, nur einen Teil der Gesamtmenge aller Variablen ausmacht, die für die Veränderung der abhängigen Variablen verantwortlich sind (Abb. 5–8). Die Effekte der übrigen Einflussgrößen (Störfaktoren) sind nicht kontrolliert und gehen als „Zufallsfehler“ in das Endergebnis ein. Die Größe dieses Fehlers kann man durch das Kontrollgruppenverfahren analysieren. Obendrein kann versucht werden, durch statistische Analysen die wesentlichen Faktoren für den Zufallsfehler herauszufinden und diese gegebenenfalls in neuen Experimenten zu überprüfen.
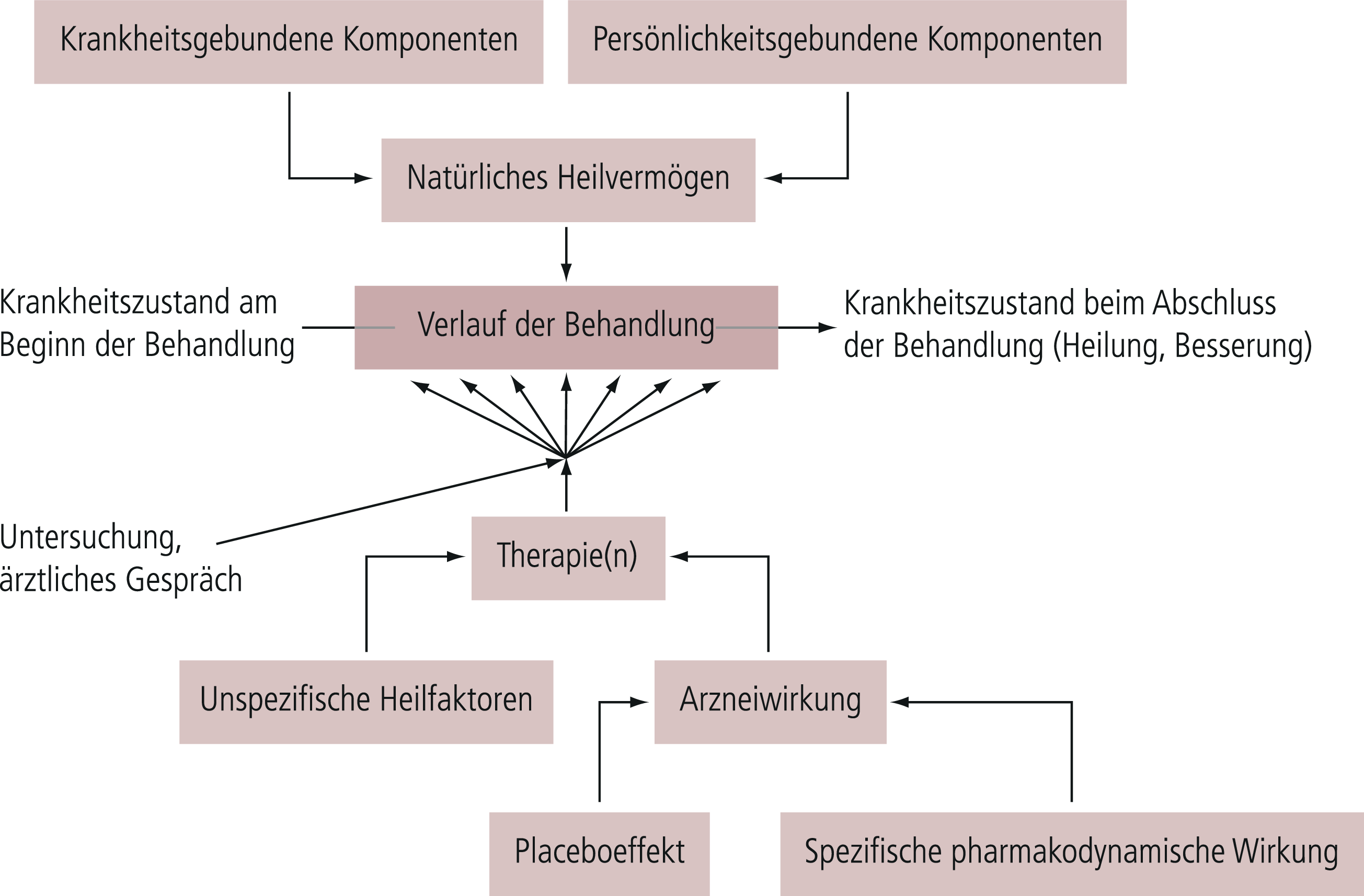
Abb. 5–8. Einflussgrößen bei Therapiestudien (mod. nach [13])
In der klinischen psychopharmakologischen Forschung wird üblicherweise eine Abstraktion von den anderen Einflussgrößen zugunsten der Wirkvariablen, dem Pharmakon (ggf. in unterschiedlichen Dosierungen), vollzogen. Dem entspricht die Bevorzugung univariater experimenteller Studien, bei denen die anderen Einflussgrößen nicht variiert oder manipuliert werden. Zumeist werden die Ergebnisse solcher univariater klinisch-psychopharmakologischer Studien lediglich sekundär unter dem Aspekt ausgewertet, korrelative Zusammenhänge zwischen bestimmten anderen Einflussgrößen und Therapieresultaten herzustellen. Dabei interessieren beispielsweise Zusammenhänge mit der Serumkonzentration des Medikaments oder mit anderen, durch das Medikament induzierten Veränderungen, beispielsweise Änderung der Konzentration von Transmittermetaboliten oder von neuroendokrinologischen Parametern.
Ergeben sich mehrere therapierelevante Faktoren bzw. sind von vornherein mehrere dieser Faktoren bekannt, kann man versuchen, gleichzeitig den Effekt dieser einzelnen Faktoren sowie die Wechselwirkung zwischen den Faktoren abzuschätzen, indem man in einem Experiment mehrere Faktoren systematisch variiert. Eine solche multivariate Dependenzanalyse ist gegenüber der oben beschriebenen univariaten Dependenzanalyse wesentlich informationsreicher. Allerdings setzt sie erheblich größere Fallzahlen voraus, insbesondere wenn man möglichst viele für die Therapie bei psychischen Krankheiten relevante Faktoren einbeziehen will.
Reduziert man in einem multivariaten Design die Einflussgrößen von vornherein auf wenige, entsteht das Problem, dass in den einzelnen Zellen zwar eine homogene Verteilung der als relevant angesehenen Faktoren, aber eine inhomogene Verteilung der nicht als relevant angesehenen Faktoren vorliegt und diese eventuell das Ergebnis wesentlich beeinflussen. Wegen dieser Probleme wird in der klinisch-psychopharmakologischen Therapieforschung immer wieder auf das Modell univariater Dependenzanalysen unter der Hilfs-Hypothese zurückgegriffen, dass alle anderen Faktoren im Vergleich zu der untersuchten Variablen vernachlässigt und in weiteren statistischen Auswertungsschritten hinsichtlich ihrer Relevanz beurteilt werden.
Evaluation psychotherapeutischer Verfahren
Obwohl eigentlich nicht zum Thema gehörend, sei doch dieses Thema kurz gestreift, um Gemeinsamkeiten, aber auch Unterschiede, der beiden Forschungsbereiche zu skizzieren.
Die Psychotherapieforschung – insbesondere im Bereich der kognitiven Psychotherapie und analoger Verfahren – bemüht sich heutzutage, den dargestellten Methoden-Idealen der Therapieforschung wie sie am Beispiel der klinisch-psychopharmakologischen Therapieforschung dargestellt wurden, gerecht zu werden (siehe dazu detailliertere Ausführungen in [13]). Einschränkungen ergeben sich vor allem dadurch, dass es schwierig ist, Placebo-Bedingungen zu realisieren, sodass meistens mit unzureichenden Hilfskonstruktionen gearbeitet werden muss. Auch eine Verblindung der Therapiemethoden ist nicht möglich, geschweige denn ein doppelblinder Ansatz. Die Hilfsstrategie, „blinde Rater“ einzusetzen, kann dieses Problem nicht suffizient lösen, da für den sachkundigen Experten die Hintergründe zumindest partiell erkennbar sind. Damit fallen wichtige Prinzipien, die in der psychopharmakologischen Therapieforschung als prinzipiell angesehen werden, weg. Dies sollte bei vergleichenden Analysen der Wirksamkeit von Psychopharmaka und von Psychotherapie kritisch betrachtet werden, insbesondere auch bei vergleichenden Metaanalysen von Psychopharmaka- und Psychotherapie-Studien.
Offenbar der Vergangenheit angehörig sind Studiendesigns mit weniger methodischer Stringenz, beispielsweise das Einzelgruppenverfahren. Nachteil dieses Verfahrens ist die Tatsache, dass nicht bestimmbar ist, inwieweit das Therapieergebnis durch Spontanverlauf und Placebo-Effekte bedingt ist. Auch Gruppenvergleiche, bei denen die Kontrollgruppe eine Wartegruppe ist, werden kaum noch praktiziert, da die geringe wissenschaftliche Aussagekraft bekannt ist. Auch das frühere Interesse an Einzelfallstudien, unter anderem auch solchen mit experimentellem Design und anspruchsvollen statistischen Analysemethoden wie beispielsweise der Zeitreihenanalyse, gehört anscheinend der Vergangenheit an.
Metaanalysen und ihr Stellenwert
Seit Einführung der Forderung nach klar definierten Evidenzbelegen im Gesundheitssystem und der damit einhergehenden Einführung der evidenzbasierten Medizin (EBM) haben Metaanalysen einen hohen Stellenwert in der Wirksamkeitsbeurteilung bekommen. Institutionen wie die Cochrane Collaboration in London und ihr deutscher Ableger in Freiburg sowie das weltweite Cochrane Netzwerk machen dies besonders deutlich. Viele evidenzbasierte Leitlinien, auch die deutschen S3-Leitlinien, sehen die aus Metaanalysen abgeleitete Evidenz als besonders hochrangig und als Beleg für den höchsten Evidenzgrad an (siehe die ausführliche Darstellung in [11]).
Die Zulassungsbehörden sehen die Ergebnisse aus Metaanalysen nur als sekundären Wirksamkeitsbeleg und folgen primär dem experimentellen Ansatz (Prinzip der Falsifizierung/Verifizierung von Ergebnissen) der Einzelstudien, also Zählung und Gewichtung der Ergebnisse der Einzelstudien. Dies ist auch die Ausgangsbasis der Leitlinien der World Federation of Societies of Biological Psychiatry (WFSBP) sowie der Leitlinien des International College of Psychopharmacology (CINP).
Metaanalysen kombinieren in statistischer Weise die Ergebnisse der für eine spezifische Fragestellung vorhandenen und als methodisch adäquat eingestuften publizierten Studien und, wenn möglich, auch die Ergebnisse der nicht publizierten Studien in quantitativer Weise.
Es gibt auch Metaanalysen, bei denen die Rohdaten aus Einzelstudien zusammengefasst werden. Hier sollte man besser von „pooled analyses“ sprechen. Diese kann man unterscheiden in solche, bei denen die zusammenfassende Auswertung mehrerer Studien prospektiv geplant war, und solche, bei denen das nicht der Fall war. Bei „pooled analyses“ besteht gegebenenfalls die Gefahr, dass nichtsignifikante Ergebnisse der Einzelstudien auf diese Weise verschleiert werden (sollen). Dies ist auch eine potenzielle Gefahr der klassischen Metaanalyse.
Als Ergebnis der Metaanalyse (Abb. 5–9) resultiert eine Effektgröße („effect size“), die beispielsweise in der Therapieforschung den quantitativen Unterschied zwischen den zwei verglichenen Therapien darstellt (z. B. [8, 10]).
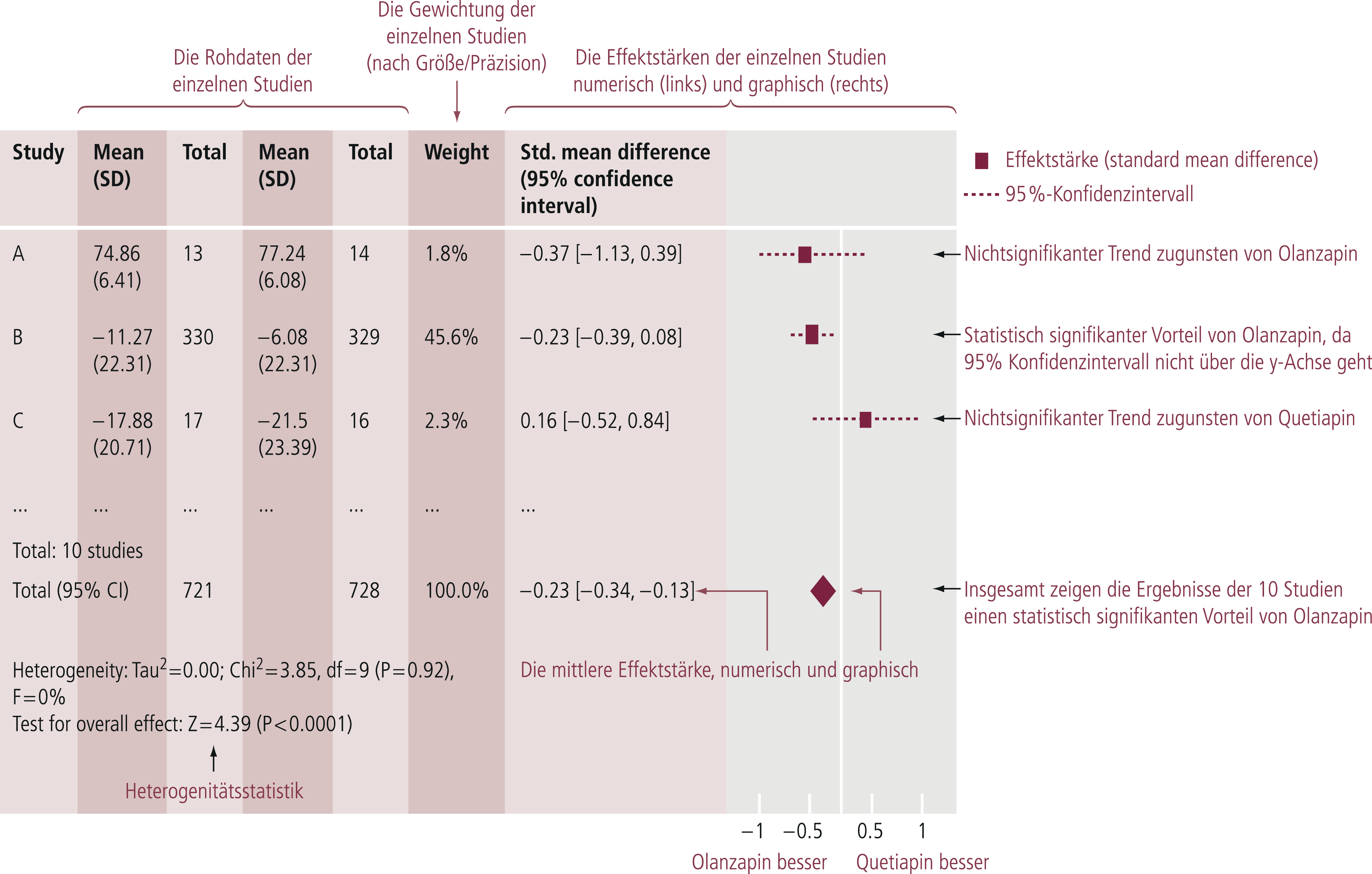
Abb. 5–9. Erklärung der Interpretation eines metaanalytischen Forest-Plots, Beispiel Wirksamkeit (Summenscore der Positive and Negative Syndrome Scale) Olanzapin vs. Quetiapin bei akuter Schizophrenie (mod. nach [11])
Die Durchführung von Metaanalysen wie auch der Vergleich von Effektgrößen setzt voraus, dass die Daten aus vergleichbaren Grundgesamtheiten stammen.
Diese Voraussetzung ist meistens bestenfalls approximativ erfüllt, da die verschiedenen berücksichtigten Studien zum Teil auf unterschiedlichen Designs mit zum Teil unterschiedlichen Rahmenbedingungen basieren (z. B. in Bezug auf Setting-Variablen, Ein- und Ausschlusskriterien, Vorbehandlung, Begleitmedikation). Metaanalysen versuchen diesen Einflussgrößen sekundär durch zusätzliche Analysen wie Sensivitätsanalysen und Subgruppenanalysen gerecht zu werden. Wichtig ist, dass möglichst alle hinsichtlich der Fragestellung relevanten Studien eingeschlossen werden. Dies ist aufgrund von Publikationsbias, die es sowohl in der Psychopharmaka-Forschung [20] wie auch der Psychotherapie-Forschung [8] gibt, allerdings oft nicht zu realisieren.
Wichtig ist zu beachten, dass Metaanalysen nicht per se eine höhere Erkenntnisebene oder Erkenntnisqualität darstellen, sondern lediglich eine statistische Zusammenfassung von Studienergebnissen sind [18]. Sie können insbesondere nicht besser sein, als die zugrunde liegenden Studien. Die Ergebnisse bedürfen wegen möglicher methodenbedingter Artefakte, die unter anderem durch das jeweils gewählte statistische Verfahren der Metaanalyse wie insbesondere auch durch die Einbeziehung bzw. den Ausschluss von Studien resultieren, kritischer Interpretation [9, 17].
Bei klassischen Metaanalysen kann nur die „direkte Evidenz“, also beispielsweise aus allen randomisierten Studien, die Medikament A mit Medikament B bzw. A mit C in einer bestimmten Indikation verglichen haben, einbezogen werden. Stehen aber viele Medikamente für diese Indikation zur Verfügung, gibt es meistens nicht für alle Medikamente solche direkten Vergleiche. Die seit etwa einem Jahrzehnt in der Psychopharmakologie eingeführten Netzwerk-Metaanalysen schließen diese Lücke, indem sie B vs. C indirekt aus A vs. B und A vs. C („indirekte Evidenz“) ableiten (Abb. 5–10).
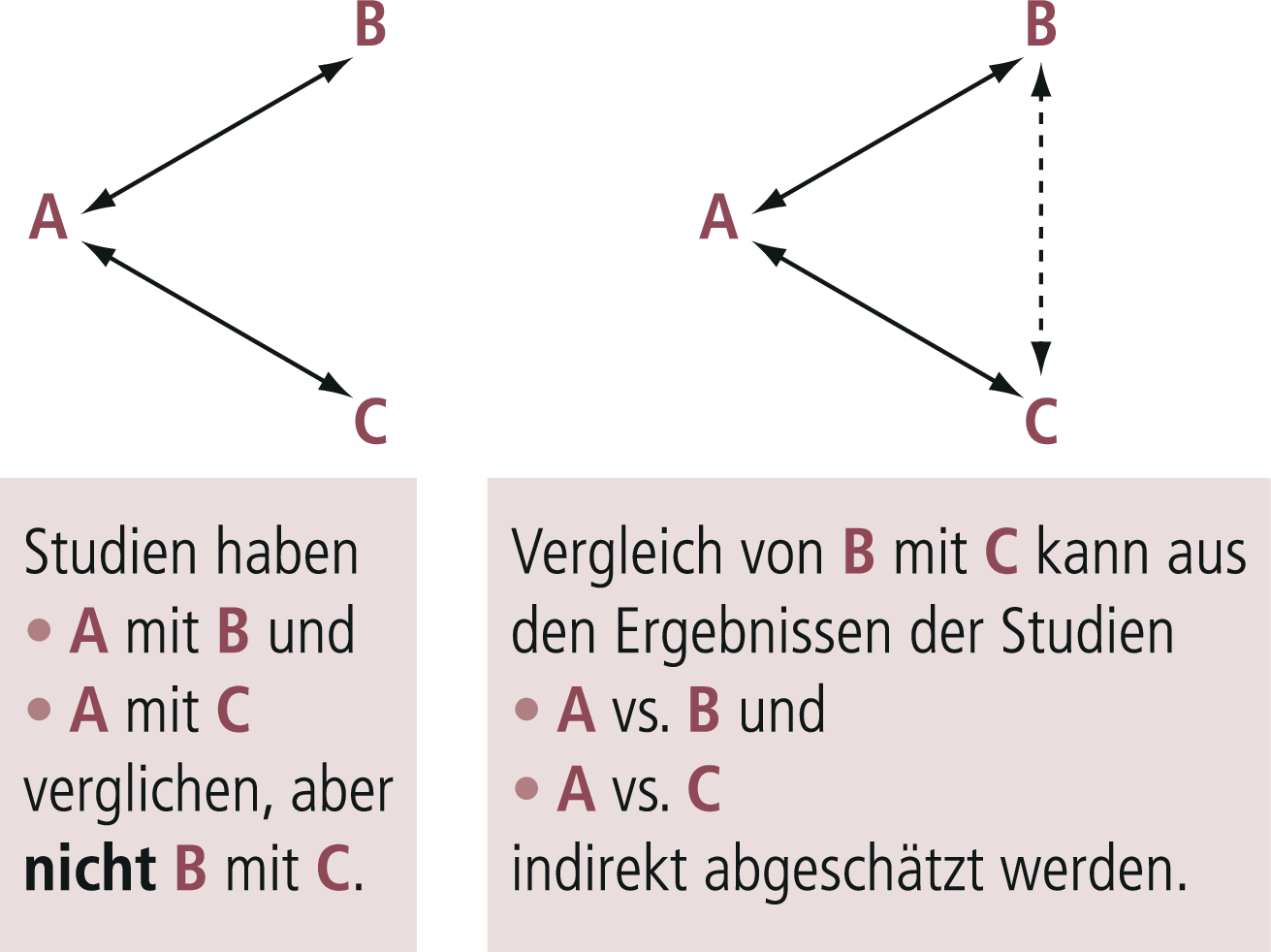
Abb. 5–10. Prinzip der indirekten Evidenz von Netzwerkmetaanalysen. Das Prinzip von Netzwerkmetaanalysen besteht darin, dass direkte Evidenz (d. h. Evidenz, die aus Studien abgeleitet wird, in denen die Medikamente direkt miteinander verglichen wurden, hier A vs. B und A vs. C) mit indirekter Evidenz kombiniert werden kann (hier B vs. C aus A vs. B und A vs. C geschätzt) (mod. nach [11]).
Der Vorteil dieses Verfahrens besteht darin, dass die gesamte Evidenz über eine Fragestellung verwendet werden kann, und dass entsprechende Wirksamkeits-Hierarchien aufgestellt werden können. Der Nachteil besteht darin, dass das Heranziehen indirekter Evidenz eine zusätzliche Annahme erfordert, nämlich dass die indirekte Evidenz valide ist. Diese Methode ist noch stör-/biasanfälliger als übliche Metaanalysen, deren methodologische Problematik immer wieder thematisiert wurde. Als Beispiel sei hier die erste Netzwerk-Metaanalyse der Antidepressiva-Studien Cipriani et al. [6] erwähnt, der inzwischen kürzlich eine weitere aktualisierte Netzwerk-Metaanalyse gefolgt ist [5]. Die riesige Datenbasis (552 randomisierte Kontrollgruppenstudien [„randomised controlled trials“, RCT], 116 477 Studienteilnehmer in die Analyse einbezogen; primäre Ausgangsbasis 28 552 Studien!) ist bemerkenswert.
Wichtig ist, sich immer wieder vor Augen zu halten, dass Metaanalysen, auch Netzwerk-Metaanalysen, keinen höheren Grad der Erkenntnis darstellen, sondern lediglich eine statistische Zusammenfassung von Daten auf einfache Kenngrößen („Effektstärke“, „effect size“). Deshalb können sie auch keinesfalls die eigentliche empirische Forschung ersetzen. Sie können aber sinnvolle und prüfbare Hypothesen für die empirische Forschung aufstellen. Auch kann ihr Ergebnis mit früheren Metaanalysen und vor allem mit allgemeiner klinischer Erfahrung verglichen werden.
Zulassungsvoraussetzungen aus Sicht der Zulassungsbehörden
Die Zulassungsvoraussetzungen für Arzneimittel sind in den letzten Jahrzehnten europaweit mit dem Ziel harmonisiert worden, qualitativ hochwertige, wirksame und sichere Arzneimittel bedarfsgerecht in der Europäischen Union zur Verfügung zu haben. Vor der Zulassung sind den Zulassungsbehörden dazu die Daten aus klinischen Prüfungen vorzulegen, die nach den oben dargelegten Prinzipien der empirischen Forschung erhoben und ausgewertet wurden. Die Genehmigung und Überwachung dieser klinischen Prüfungen von Arzneimitteln erfolgt dabei durch die nationalen Zulassungsbehörden, wie das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) in Deutschland. Die Verordnung (EU) Nr.536/2014 löst nach einer Übergangszeit und Einrichtung eines einheitlichen elektronischen Einreichungsportals mit angeschlossener Datenbank die bisherigen Regelungen voraussichtlich im Jahr 2020 ab. Ziel der neuen Regelung ist eine stärkere Harmonisierung und Erleichterung für die Durchführung multizentrischer und multinationaler klinischer Prüfungen in Europa [19]. Für die Zulassung und Postmarketing-Überwachung ist ein komplexes Netzwerk aus nationalen und europäischen Behörden zuständig, hier ist vor allem die European Medicines Agency (EMA) zu erwähnen. Dabei sind bis zur Zulassungsentscheidung im Wesentlichen zwei Verfahrensstränge zu unterscheiden: das zentrale Verfahren der EMA – dann gilt die Zulassung verbindlich europaweit – und die dezentralen Zulassungsverfahren (dezentrales Verfahren; Verfahren der gegenseitigen Anerkennung), bei denen die Zulassung nur national oder in bestimmten europäischen Mitgliedsstaaten gilt. Innovative Arzneimittel, das heißt Arzneimittel mit neuen Wirkstoffen oder für wichtige neue Anwendungsgebiete, werden aber praktisch ausschließlich im zentralen Verfahren zugelassen. Dies gilt auch für psychiatrische und neurologische Indikationsgebiete. Dabei führt und koordiniert die EMA das Verfahren verwaltungstechnisch, die wissenschaftliche Bewertung der eingereichten Studienunterlagen wird jedoch von den Experten der nationalen Zulassungsbehörden, die unter Führung der EMA im europäischen Netzwerk zusammenarbeiten, erstellt. Eine konsentierte oder durch die Mehrheit bestimmte europäische Position wird im Ausschuss für Humanarzneimittel („Committee for Medicinal Products for Human Use“, CHMP) der EMA erarbeitet, in den jedes Mitgliedsland der EU ein stimmberechtigtes Mitglied entsendet. Die von der Mehrheit des CHMP angenommene Bewertung des Nutzens und der Risiken eines Arzneimittels ist die wissenschaftliche Grundlage für dessen Zulassung (oder Ablehnung) durch die Europäische Kommission.
Die Wirksamkeit [3] ist in der Regel durch prospektive, doppelblinde und randomisierte Studien im Vergleich mit einer geeigneten Kontrollgruppe zu belegen (siehe Teil 1 dieser Darstellung [14]!).
Um die Zulassungsvoraussetzungen zu harmonisieren, aber auch um Hilfestellungen für die Durchführung entsprechender klinischer Prüfungen zu geben, sind von den Experten der nationalen Zulassungsbehörden in indikationsspezifischen Arbeitsgruppen (z. B. für den psychopharmakologischen Bereich in der „Central Nervous System Working Party“ bei der EMA) Leitlinien zur Durchführung klinischer Prüfungen publiziert worden. In diesen finden sich konkrete Angaben zu geeigneten Studienpopulationen, Studiendesigns, Erhebungsinstrumenten, primären und sekundären Endpunkten, Studiendauer, statistischen Auswertestrategien usw. und sie berücksichtigen die oben beschriebenen methodischen Standards (Übersicht in [13]; aktueller Link: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/clinical-efficacy-safety/clinical-efficacy-safety-nervous-system). Die EMA-Leitlinien sollen den pharmazeutischen Unternehmen die Planung ihrer Entwicklungsprogramme erleichtern, sind aber gleichzeitig für methodisch interessierte Kliniker und Prüfärzte von Bedeutung, um ein vertieftes Verständnis für diese Fragestellungen und deren adäquate Umsetzung zu bekommen.
In der Regel werden für die Einschlusskriterien die Diagnosekriterien des diagnostischen und statistischen Manuals psychischer Störungen (DSM) in der jeweils aktuellen Version empfohlen, mit nachvollziehbaren Begründungen kann davon aber auch abgewichen werden.
Wenn ein Arzneimittel wirksam ist und sicher angewendet werden kann, erhält es mit der Zulassung den Marktzugang. Neue Arzneimittel müssen gegenüber früher schon zugelassenen Arzneimitteln nach den Vorgaben des Arzneimittelgesetzes aber nicht überlegen sein, hier soll bewusst auch die Verfügbarkeit von mehreren Therapiealternativen mit beispielsweise unterschiedlichen Nebenwirkungsprofilen möglich sein.
Ein mehrarmiges Studiendesign mit Placebo, aktiver Kontrolle und neuer Prüfsubstanz stellt aus regulatorischer Sicht den bestmöglichen Kompromiss der verschiedenen beschriebenen Studiendesigns dar, da deren Vorteile unter Minimierung der Nachteile berücksichtigt werden. Der Placebo-Arm ermöglicht die Bestimmung der Assay-Sensitivität in der klinischen Prüfung und die Abschätzung der Effektstärke des Prüfmedikaments. Darüber hinaus erlaubt dieses Design die Einschätzung der klinischen Relevanz der Studienergebnisse der neuen Prüfsubstanz im Vergleich zu Placebo und zum etablierten Standardpräparat, auch lässt sich das Nebenwirkungsprofil der Prüfsubstanz Placebo-bereinigt gegenüber der Standardreferenz vergleichen.
Die Berücksichtigung dieser Prinzipien ist in Deutschland aktuell auch wegen des Arzneimittelmarktneuordnungsgesetzes (AMNOG) noch wichtiger geworden. Wenige Monate nach Markteinführung ist beim Gemeinsamen Bundesausschuss (G-BA) ein Dossier mit Studiendaten einzureichen, das einen Zusatznutzen des neuen Arzneimittels im Vergleich zu etablierten Therapiestandards nachweisen muss. Hierfür wird vom G-BA eine sogenannte „zweckmäßige Vergleichstherapie“ festgelegt. Basis der Beurteilung durch den G-BA sind in der Regel auch die für die Zulassung durchgeführten Phase-III-Studien, die gegebenenfalls um weitere klinische Prüfungen ergänzt werden müssen. Während die Zulassung europaweit durch die Arzneimittelgesetze harmonisiert ist, gilt dies für die Zusatznutzenbewertung noch nicht; die Vorgaben für diese Bewertung kommen in Deutschland aus dem nationalen SGB V und sind dort in § 35a festgelegt. Entscheidend sind dabei neben dem Nachweis der Wirksamkeit für die Zusatznutzenbewertung die Berücksichtigung patientenrelevanter Endpunkte wie Mortalität, Morbidität und Lebensqualität. BfArM und G-BA bieten hierzu eine gemeinsame Beratung für Sponsoren klinischer Prüfungen an [4]. Aber auch für die Zusatznutzenbewertung gibt es Bestrebungen der EU-Kommission, zumindest die Methodik solcher Ansätze zu harmonisieren.
Interessenkonflikterklärung
Keine Interessenkonflikte.
Literatur
1. Baldwin DS, Loft H, Dragheim M. A randomised, double-blind, placebo controlled, duloxetine-referenced, fixed-dose study of three dosages of Lu AA21004 in acute treatment of major depressive disorder (MDD). Eur Neuropsychopharmacol 2012;22:482–91.
2. Boulenger JP, Loft H, Florea I. A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. J Psychopharmacol 2012;26:1408–16.
3. Broich K. Klinische Prüfungen mit Antidepressiva und Antipsychotika. Bundesgesundheitsblatt 2005;48:541–7.
4. Broich K, Lobker W, Schulte A, Beinlich P, et al. Anforderungen an Zulassung und Nutzenbewertung von Arzneimitteln – Regulatorische Aspekte und Erfahrungen. Nervenarzt 2016;87:376–85.
5. Cipriani A, Furukawa TA, Salanti G, Chaimani A, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet 2018;391:1357–66.
6. Cipriani A, Furukawa TA, Salanti G, Geddes JR, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009;373:746–58.
7. Cohen J. Statistical power analysis for the behavioral sciences. 2. Auflage. Hillsdale, NJ, USA: Lawrence Earlbaum Associates, 1988.
8. Cuijpers P, Berking M, Andersson G, Quigley L, et al. A meta-analysis of cognitive-behavioural therapy for adult depression, alone and in comparison with other treatments. Can J Psychiatry 2013;58:376–85.
9. Fountoulakis KN, Veroniki AA, Siamouli M, Moller HJ. No role for initial severity on the efficacy of antidepressants: results of a multi-meta-analysis. Ann Gen Psychiatry 2013;12:26.
10. Leucht S, Kissling W, Davis JM. How to read and understand and use systematic reviews and meta-analyses. Acta Psychiatr Scand 2009;119:443–50.
11. Leucht S, Möller H-J. Evidenzbasierung und leitliniengeschützte Therapie in der Psychiatrie. In: Möller H-J, Laux G, Kapfhammer H-P (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Berlin, Heidelberg: Springer-Verlag, 2017.
12. Mallinckrodt CH, Raskin J, Wohlreich MM, Watkin JG, et al. The efficacy of duloxetine: a comprehensive summary of results from MMRM and LOCF_ANCOVA in eight clinical trials. BMC Psychiatry 2004;4:26.
13. Möller H-J, Broich K. Prinzipien der Methodik empirischer Forschung in der Psychiatrie. In: Möller H-J, Laux G, Kapfhammer H-P (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Berlin, Heidelberg: Springer-Verlag, 2017:463–90.
14. Möller H-J, Broich K. Methodik klinischer psychopharmakologischer Therapieforschung (Teil I): Allgemeine Grundlagen, 4-Phasen-Modell, Design klinischer Prüfungen. Psychopharmakotherapie 2018;25:251–7.
15. Moller HJ. Do effectiveness („real world“) studies on antipsychotics tell us the real truth? Eur Arch Psychiatry Clin Neurosci 2008;258:257–70.
16. Moller HJ. Isn‘t the efficacy of antidepressants clinically relevant? A critical comment on the results of the metaanalysis by Kirsch et al. 2008. Eur Arch Psychiatry Clin Neurosci 2008;258:451–5.
17. Moller HJ, Maier W. Probleme der „evidence-based medicine“ in der Psychopharmakotherapie: Problematik der Evidenzgraduierung und der Evidenzbasierung komplexer klinischer Entscheidungsprozesse [Problems of evidence-based medicine in psychopharmacotherapy: problems of evidence grading and of the evidence basis for complex clinical decision making]. Nervenarzt 2007;78:1014–27.
18. Moller HJ, Maier W. Evidence-based medicine in psychopharmacotherapy: possibilities, problems and limitations. Eur Arch Psychiatry Clin Neurosci 2010;260:25–39.
19. Nickel L, Seibel Y, Frech M, Sudhop T. Änderungen des Arzneimittelgesetzes durch EU-Verordnung zu klinischen Prüfungen. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 2017;60:804–11.
20. Turner EH, Matthews AM, Linardatos E, Tell RA, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med 2008;358:252–60.
21. Volz H-P, Kasper S, Möller H-J. Psychopharmakotherapie – klinisch-empirische Grundlagen. In: Möller H-J, Laux G, Kapfhammer H-P (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Berlin, Heidelberg: Springer-Verlag, 2017: 795–842.
Herrn Prof. Dr. med. Dipl.-Psych. Gerd Laux zum 70. Geburtstag in großer Dankbarkeit für eine fast lebenslange erfolgreiche Zusammenarbeit vom Erstautor gewidmet.
Prof. Dr. med. Dr. h. c. mult. Hans-Jürgen Möller, Ehemaliger Direktor der Klinik für Psychiatrie und Psychotherapie, Nußbaumstraße 7, 80336 München, E-Mail: hans-juergen.moeller@med.uni-muenchen.de
Prof. Dr. Karl Broich, Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn
Methodology of clinical treatment research on psychopharmaceuticals (part II): sample selection, statistical methods, meta-analyses, regulatory framework
This paper presents the importance of using appropriate methods to select samples and of testing hypotheses with suitable statistical methods. It explains special evaluation procedures, including the observed case analysis and last observation carried forward analysis, and describes the value of various meta-analytical approaches. The paper ends by describing the regulatory framework.
Key words: sample selection, statistical methods, meta-analysis, regulatory framework
Psychopharmakotherapie 2018; 25(06):304-312