Isabella Stuckart, Timo Siepmann, Heinz Reichmann und Kristian Barlinn, Dresden
Schlaganfall und Depression
In Deutschland erleiden jährlich etwa 196000 Menschen einen ersten Schlaganfall und 66000 Menschen ein Schlaganfallrezidiv [22]. Der Schlaganfall steht seit Jahren an dritter Stelle der Todesursachen in Deutschland. Zudem stellt dieses Krankheitsbild die häufigste Ursache erworbener Behinderungen im Erwachsenenalter dar, woraus neben individuellen Einschränkungen auch erhebliche invaliditätsbezogene gesundheitsökonomische Folgen resultieren. Während in den vergangenen Jahrzehnten der wissenschaftliche Fokus vorrangig auf der Akuttherapie des Schlaganfalls lag, rücken auch die gesundheitlichen Folgen der Erkrankung und Schlaganfallnachsorge zunehmend in den Mittelpunkt wissenschaftlicher und gesundheitspolitischer Interessen [5].
Ebenso wie der Schlaganfall zählt in Deutschland auch die Depression zu den häufigen Krankheitsbildern von hoher medizinischer und gesundheitsökonomischer Relevanz [6]. Über einen Zeitraum von 12 Monaten findet sich eine Depression bei etwa 6% der Bevölkerung. Unabhängig von den aus einer Depression resultierenden immensen sozialpsychiatrischen Konsequenzen stellt die Erkrankung auch einen Risikofaktor für körperliche Erkrankungen, zum Beispiel den Schlaganfall, dar [4]. Patienten mit Schlaganfall erleiden überzufällig häufig eine Depression [19].
Diese Übersichtsarbeit beschäftigt sich mit dem Krankheitsbild der Post-Schlaganfall-Depression (PSD), also einer nach Schlaganfall auftretenden depressiven Störung.
Epidemiologie der Post-Schlaganfall-Depression
Die PSD ist eine häufige und schwere Komplikation des Schlaganfalls [42, 49]. Einer zuletzt im Jahr 2014 aktualisierten Metaanalyse prospektiver Beobachtungsstudien zufolge lässt sich bei etwa 31% aller Überlebenden zu einem unbestimmten Zeitpunkt nach einem Schlaganfall eine depressive Störung nachweisen [19]. Die Häufigkeit scheint mit 25% nach dem ersten Jahr etwas abzunehmen, wobei nach fünf Jahren noch immer 23% aller Schlaganfallpatienten unter einer PSD leiden. Dabei unterscheidet sich die Häufigkeit der PSD nicht zwischen ischämischen und hämorrhagischen Schlaganfällen [15].
Post-Schlaganfall-Depression als Prädiktor für ungünstiges Outcome
Unabhängig von demographischen Faktoren und Schlaganfallschwere ist die PSD mit einer erhöhten Mortalität assoziiert [3]. Ergebnisse aus dem South London Stroke Register weisen auf ein um 34% erhöhtes Risiko hin, innerhalb von fünf Jahren zu versterben, wenn zum Zeitpunkt von drei Monaten nach Schlaganfall eine PSD vorliegt. Gleichfalls kann eine PSD die funktionelle Rekonvaleszenz nach einem Schlaganfall beeinträchtigen, sodass Patienten mit einer frühen Entwicklung einer PSD nach fünf Jahren einen höheren Grad an Behinderung aufweisen als solche ohne PSD [27]. Inwieweit dies auf eine eingeschränkte Mitarbeit während der Rehabilitation, kognitive Einbußen oder eine verminderte neuronale Plastizität zurückzuführen ist, bedarf weiterer wissenschaftlicher Klärung [42, 49]. Die Lebensqualität nach einem Schlaganfall ist zudem, unabhängig von der Schwere der Behinderung, durch das Vorliegen einer PSD beeinträchtigt. Auch ist das kumulative Risiko für ein Schlaganfallrezidiv innerhalb von 12 Jahren nach einem Schlaganfall bei Patienten mit PSD im Vergleich zu Patienten ohne PSD um 68% erhöht [39].
Vorhersage einer Post-Schlaganfall-Depression
Die Identifikation von Risikomarkern, die eine PSD bei Patienten mit akutem Schlaganfall vorhersagen, kann dem behandelnden Arzt helfen, Risikopatienten frühzeitig zu erkennen und gegebenenfalls prophylaktisch zu behandeln. In der Literatur finden sich mehrere Risikomarker, die eine PSD mit variabler Verlässlichkeit vorhersagen [12, 27, 39, 49]. Hierzu zählen vor allem die Schlaganfallschwere und das Ausmaß einer nachfolgenden funktionellen Behinderung. Entsprechend eines validierten Prädiktionsmodells weisen akute Schlaganfallpatienten, die sich aufgrund eines neurologischen Defizits nicht selbstständig an- und auskleiden können (gemäß Barthel-Index Item 4), ein um 57% erhöhtes Risiko auf, innerhalb von acht Wochen eine PSD zu entwickeln, im Vergleich zu Patienten, denen diese Fähigkeit geblieben ist [12]. Zu den weiteren etablierten Risikomarkern zählen Depression oder kognitive Beeinträchtigung in der Vorgeschichte. So hatten in einer Studie Patienten mit vorbestehender kognitiver Beeinträchtigung ein 3-fach erhöhtes Risiko, innerhalb von einem Monat nach Schlaganfall eine PSD auszubilden [39]. Soziale Deprivation, weibliches Geschlecht und eine hohe Last vaskulärer Risikofaktoren scheinen ebenfalls eine PSD vorherzusagen.
Ätiologie der Post-Schlaganfall-Depression
Die zur Entstehung einer PSD führenden Pathomechanismen sind weitgehend unverstanden [42, 49]. Es herrscht jedoch allgemeiner Konsens, dass die Ätiologie, ähnlich der Depression bei Patienten ohne Schlaganfall, als multifaktoriell anzusehen ist. Dabei werden neben psychosozialen Aspekten, zum Beispiel Versagen von Coping-Strategien und depressiven Anpassungsstörungen als Folge körperlicher oder kognitiver Beeinträchtigungen, vor allem auch neurobiologische Störungen als unmittelbare Folge der Hirnschädigung diskutiert.
Theoretische Modelle zur neurobiologischen Genese der PSD
Der Theorie nach führen durch einen Schlaganfall hervorgerufene strukturelle Schäden an den aufsteigenden monoaminergen Systemen zu einer Verminderung der serotoninergen, dopaminergen und noradrenergen Neurotransmission. Insbesondere bei einer Beteiligung der die Stimmung modulierenden limbischen bzw. frontalen und temporalen Hirnregionen kann dies mit der Manifestation einer depressiven Störung einhergehen [29, 49]. In einer klinischen Pilotstudie von Glodzik-Sobanska et al. zeigte sich magnetresonanzspektroskopisch bei Schlaganfallpatienten mit PSD eine im Vergleich zur gesunden Hemisphäre deutlich verminderte Glutamatkonzentration im anterioren cingulären Kortex, was neben dem erwähnten Monoamin-Mangel zusätzlich eine pathophysiologische Rolle der Glutamat-Neurotransmission in der Entstehung der PSD vermuten lässt [16]. Die Beobachtung, dass vornehmlich in der linken anterioren Hemisphäre lokalisierte ischämische Läsionen mit einer Depression vergesellschaftet sind, konnte in einer Metaanalyse klinischer Beobachtungsstudien nicht bestätigt werden. Ein Zusammenhang zwischen Läsionslokalisation und Inzidenz einer PSD ließ sich hier nicht aufzeigen [51].
Klinische Daten weisen zudem auf eine Assoziation zwischen einer aus zerebraler Schädigung resultierenden hypothalamisch-hypophysären Funktionsstörung und dem Auftreten einer PSD hin. Erstmals konnte ein entsprechender Zusammenhang von Aström et al. in einer Kohorte von 70 Schlaganfallpatienten beobachtet werden, nachdem ein in der Subakutphase erhöhter Postdexamethason-Spiegel von Cortisol signifikant eine PSD im Langzeitverlauf voraussagte [2]. Den Ergebnissen einer rezenten Metaanalyse zufolge ist von einem 3,28-fachen Risiko für eine PSD bei Nachweis erhöhter Cortisol-Spiegel nach Dexamethason-Suppression-Test auszugehen [38].
Tierexperimentelle Daten legen nahe, dass es im Rahmen einer zerebralen Ischämie zu einer vermehrten Differenzierung von Stammzellen im Hippocampus und in der subependymalen Zone kommt, die nachfolgend in Richtung der von einer Ischämie im Rahmen eines Schlaganfalls betroffenen Hirnareale migrieren können [29, 46]. Auch ließ sich im Tierexperiment, basierend auf der Induktion eines ischämischen Schlaganfalls durch Okklusion der Arteria cerebri media bei adulten Ratten, durch die Implantation von Stammzellen der subependymalen Zone und gleichzeitige multimodale Therapie eine verstärkte Differenzierung und Migration dieser Neurone beobachten. Dieser multimodale Ansatz resultierte in einer Verbesserung des funktionellen Outcomes [23]. Inwieweit zur Differenzierzung und Migration angeregte Stammzellen die Funktion zugrunde gegangener Neuronen übernehmen können, ist jedoch unklar. Die adulte Neurogenese scheint aber auch eine Rolle in der Prävention und der Rekonvaleszenz einer Depression zu spielen. Umgekehrt hemmen die einer Depression zugrunde liegenden Mechanismen die Stimulation der adulten Neurogenese, was sich wiederum nachteilig auf die Erholung nach einem Schlaganfall und sekundär auf die Akzentuierung depressiver Störung auswirken könnte. In welchem Umfang diese aus tierexperimentellen Studien abgeleiteten bidirektionalen Beziehungen zwischen Schlaganfall, Depression und adulter Neurogenese eine pathophysiologische Rolle in der klinischen Entstehung einer PSD spielen, erfordert weitere wissenschaftliche Prüfung [29]. Interessanterweise erwirkt eine pharmakotherapeutische Intervention mit Antidepressiva einen günstigen Effekt auf das Infarktvolumen, möglicherweise auf einer Stimulation der adulten Neurogenese basierend. In einer rezenten Metaanalyse aus 29 tierexperimentellen Publikationen, in denen 12 unterschiedliche Antidepressiva erprobt wurden, fand sich im Vergleich zu Kontrollen eine Reduktion des Infarktvolumens um 27%, wobei dieser Effekt unabhängig von dem verwendeten Wirkstoff beziehungsweise der Substanzklasse zu beobachten war [33].
Neben den vorgestellten Theorien zur neurobiologischen Genese werden zudem einem Mangel an Wachstumsfaktor BDNF (Brain-derived neurotrophic factor), einer globalen Reduktion des zerebralen Blutflusses sowie einer Reduktion des Amygdala-Volumens eine Bedeutung in der Entstehung einer PSD zugesprochen [38, 49].
Vaskuläre Depression
Im Kontext ätiologischer Überlegungen soll der Vollständigkeit halber auf die erstmals im Jahr 1997 von Alexopoulos et al. generierte Hypothese zur vaskulären Depression eingegangen werden [47]. Demnach stellten sich bei Patienten mit spätem Beginn einer Depression (nach dem 65. Lebensjahr) in der zerebralen Bildgebung häufiger chronische mikrovaskuläre Veränderungen dar als bei Patienten mit frühem Beginn einer Depression. So fand sich in einer Metaanalyse eine etwa 4-fach erhöhte Wahrscheinlichkeit für das Vorhandensein von chronischen mikrovaskulären Veränderungen bei Patienten mit später im Vergleich zu Patienten mit früher Manifestation einer Depression [21]. Auch legen postmortale Studien einen Zusammenhang nahe [25]. Somit könnten nicht nur eine einem Schlaganfall unmittelbar folgende Hirnschädigung, sondern auch chronische mikrovaskuläre Veränderungen (v.a. unter Beteiligung der kortiko-striato-pallido-kortikalen Leitungsbahnen), häufig klinisch stumm verlaufend, das Risiko einer Depression erhöhen, wobei ähnliche zugrunde liegende Pathomechanismen infrage kommen. Allerdings nehmen chronische mikrovaskuläre Veränderungen generell mit dem Alter zu, weswegen es sich auch um eine koinzidente Beobachtung bei Patienten mit Spätbeginn einer Depression handeln könnte [41].
Diagnostik der Post-Schlaganfall-Depression
Trotz enormen Fortschritten in der Akutversorgung von Schlaganfallpatienten erfolgt in der klinischen Praxis selten ein systematisches Screening auf das Vorliegen einer PSD. Zu erklären ist dies mit der Tatsache, dass die PSD erst seit wenigen Jahren als neue Entität wahrgenommen wird [14, 42]. Eine Erkennung gestaltet sich zudem durch eine erschwerte Abgrenzung von schlaganfallbezogenen Symptomen schwierig. So kann zum Beispiel eine aphasische oder somatomotorische Störung mit Beeinträchtigung der Mobilität eine schwere Antriebsstörung maskieren. Zusätzlich kann eine Abgrenzung von demenziellen Störungen erschwert sein, da vor allem ältere Patienten mit einem Schlaganfall zusätzlich an einem demenziellen Syndrom erkrankt sein können.
Grundsätzlich unterscheidet sich die Diagnosestellung der PSD nicht von einer nicht schlaganfallbezogenen Depression, wobei die operationalen Diagnosekriterien der internationalen Klassifikationssysteme ICD-10 bzw. DSM 5 herangezogen werden können [13]. Da sich eine PSD bereits frühzeitig in der Akutphase eines Schlaganfalls manifestieren kann, ist die Einhaltung des Zeitkriteriums dieser Klassifikationen (d.h. Bestehen der Symptome über mindestens zwei Wochen) nicht sinnvoll. Neben einer strukturierten psychiatrischen Exploration werden auch psychometrische Beurteilungsskalen in der Diagnostik der PSD angewandt. Es herrscht jedoch Unklarheit, welcher der in der Diagnostik einer Depression üblicherweise eingesetzten Selbst- bzw. Fremdbeurteilungsbögen auch Patienten mit einer PSD zuverlässig identifiziert. In einer von Meader et al. durchgeführten Metaanalyse von 24 diagnostischen Studien zur ICD-10- bzw. DSM-5-basierten Diagnostik einer PSD fanden sich optimale prädiktive Werte bei Verwendung der Center of Epidemiological Studies-Depression Scale (CES-D), Hamilton Depression Rating Scale (HDRS) und des Patient Health Questionnaire (PHQ-9), wobei die Sensitivität 75% bis 86% betrug [36, 49]. Ergänzend möchten wir darauf hinweisen, dass die HDRS primär zur Beurteilung des Schweregrads einer Depression nach erfolgter Diagnose konzipiert wurde und daher stets mit anderen Instrumenten verwendet werden sollte. Da die Beurteilung mittels Selbst- bzw. Fremdbeurteilungsbögen aufgrund von motorischen, sprachlichen oder neuropsychologischen Defiziten nicht bei allen Schlaganfallpatienten umsetzbar ist, bleibt nicht selten der alleinige klinische Eindruck, um das Vorliegen einer PSD zu vermuten. Eine Beobachtungsstudie von Carota et al. konnte in diesem Kontext zeigen, dass die Wahrscheinlichkeit für die Entwicklung einer PSD 2,66-fach erhöht ist, wenn das Pflegeteam bei betroffenen Patienten Weinen oder offensichtliche Traurigkeit wahrnimmt [8].
Behandlung der Post-Schlaganfall-Depression
Ähnlich der Depression stehen zur Behandlung einer PSD pharmakotherapeutische und nicht-pharmakotherapeutische (v.a. psychotherapeutische) Interventionen zur Verfügung. Da sich diese Übersichtsarbeit im Folgenden ausschließlich mit der Psychopharmakotherapie der PSD befasst, sei an dieser Stelle bezüglich nicht-pharmakotherapeutischer Ansätze auf die Literatur verwiesen [49].
Pharmakotherapeutische Prophylaxe der Post-Schlaganfall-Depression
Eine aktuelle Metaanalyse von acht randomisierten kontrollierten Studien (n=776) zeigte einen positiven Effekt zugunsten einer prophylaktischen Behandlung mit Antidepressiva bei Schlaganfallpatienten ohne erkennbare Depression zum Zeitpunkt des Studieneinschlusses [43]. Die untersuchten Substanzklassen umfassten selektive Serotonin-Wiederaufnahmehemmer (SSRI; Escitalopram, Fluoxetin, Sertralin), selektive Noradrenalin-Wiederaufnahmehemmer (Milnacipran), tetrazyklische Antidepressiva (Mianserin, Mirtazapin) und trizyklische Antidepressiva (Nortriptylin). Die Therapie wurde zwischen dem 1. Tag und dem 6. Monat nach Schlaganfall begonnen und für 3 bis 12 Monate fortgeführt. Das Risiko für die Entwicklung einer PSD war unter der antidepressiven Therapie im Vergleich zu Kontrollen um 66% niedriger (Odds-Ratio [OR] 0,34; 95%-Konfidenzintervall [KI] 0,02–0,53). Unter alleiniger Berücksichtigung der Studien, in denen die Therapiedauer 12 Monate betrug, fand sich eine Risikoreduktion von 69% (OR 0,31; 95%-KI: 0,18–0,56), innerhalb des ersten Jahres nach Schlaganfall an einer PSD zu erkranken. Bei isolierter Betrachtung von Studien (n=5), in denen ein SSRI verordnet wurde, fanden sich ähnliche Ergebnisse. Auch hier war unter prophylaktischer Behandlung mit SSRI das Risiko für die Entwicklung einer PSD im Vergleich zu Kontrollpatienten um 63% reduziert (OR 0,37; 95%-KI 0,22–0,61). In Bezug auf unerwünschte Arzneimittelwirkungen waren keine Unterschiede zwischen den Behandlungsgruppen zu beobachten.
Patienten mit einer aphasischen Störung oder kognitiven Beeinträchtigung wurden in allen Studien ausgeschlossen, was eine eingeschränkte Generalisierbarkeit der Ergebnisse bedingt. Zudem bestand in den inkludierten Studien eine methodische Vielfalt in Bezug auf das Studiendesign (z.B. Diagnosekriterien und Evaluationszeitraum für Depression, Zeitpunkt der Therapieinitiierung und Therapiedauer), sodass es weiterer klinischer Prüfungen hinsichtlich eines prophylaktischen Einsatzes von Antidepressiva bei Schlaganfallpatienten bedarf.
Pharmakotherapie der Post-Schlaganfall-Depression
Es existieren nur wenige randomisierte kontrollierte Studien zum Effekt einer pharmakotherapeutischen Intervention bei Patienten mit einer PSD. In einer zuletzt im Jahr 2008 aktualisierten systematischen Übersichtarbeit der Cochrane Collaboration wurden lediglich 12 Studien (n=1121) mit auswertbaren Outcome-Daten identifiziert [18]. In den inkludierten Studien wurden die trizyklischen Antidepressiva Amitriptylin und Nortriptylin, die SSRI Citalopram, Fluoxetin (n=2), Paroxetin (n=3), Reboxetin, Sertralin und Trazodon sowie das Nootropikum Aniracetam geprüft, wobei die Therapieinitiierung zwischen wenigen Tagen und 25 Monaten nach Schlaganfall erfolgte. Die Diagnose einer PSD wurde anhand eines psychiatrischen Interviews oder psychometrischer Beurteilungsskalen gestellt.
Eine Remission der PSD trat unter antidepressiver Therapie um 53% (OR 0,47; 95%-KI 0,22–0,98) häufiger auf als unter Kontrollbedingungen (z.B. Placebo oder Psychotherapie). Auch fand sich in der Aktivgruppe signifikant häufiger eine mindestens 50%ige Reduktion des Punktwerts auf den jeweils verwendeten psychometrischen Beurteilungsskalen (OR 0,22; 95%-KI 0,09–0,52). Eine Subanalyse nach bestimmten Substanzklassen (z.B. SSRI) erfolgte aufgrund einer beträchtlichen Heterogenität im Design der Studien nicht.
Eine im Jahr 2017 publizierte Netzwerk-Metaanalyse, die 12 randomisierte kontrollierte Studien (n=707; überwiegend überlappend mit der Analyse der Cochrane Collaboration) inkludierte, zielte auf einen individuellen Vergleich der verwendeten antidepressiven Wirkstoffe in Bezug auf Akzeptanz und Wirksamkeit ab [48]. Demnach bestand für Paroxetin in der Behandlung einer PSD das günstigste Verhältnis zwischen Akzeptanz und Wirksamkeit. Zu einem sehr ungünstigen Verhältnis zwischen Akzeptanz und Wirksamkeit kamen die Autoren bei metaanalytischer Betrachtung von Fluoxetin. Unter alleiniger Betrachtung der Wirksamkeit schien Reboxetin, ein selektiver Noradrenalin-Wiederaufnahmehemmer (NARI), den größten Therapieeffekt auf eine Depression im Vergleich zu Placebo zu haben. Zu bedenken bei dieser Substanzklasse ist jedoch eine noradrenerge Wirkung, die zu einer arteriellen Hypertonie führen oder diese akzentuieren kann. Bei Schlaganfallpatienten sollten im Rahmen der Sekundärprophylaxe optimierte Blutdruckwerte angestrebt und nachteilige Einflüsse auf die Blutdrucklage vermieden werden. Bezüglich der Metaanalyse ist methodisch zu erwähnen, dass zum Teil nur einzelne Studien mit Verwendung einer spezifischen Substanz berücksichtigt wurden und die maximale Behandlungszeit in den inkludierten Studien maximal 12 Wochen betrug. Dem gegenüber stehen Daten aus der Literatur, die annehmen lassen, dass die Effektivität einer Therapie der PSD von der Behandlungsdauer abhängt [48].
Obgleich die Ergebnisse dieser Metaanalysen auf einen insgesamt deutlichen günstigen Effekt der Behandlung mit Antidepressiva nach Schlaganfall hinweisen, kann aufgrund der jeweils geringen Fallzahlen sowie methodischen Vielfalt der in die Analyse einbezogenen Studien keine generelle Therapieempfehlung für eine spezifische Substanz gegeben werden. Unter Berücksichtigung eines günstigen Wirkungs-/Nebenwirkungsprofils, der vielversprechenden Hinweise auf eine prophylaktische und therapeutische Wirkung, und der im Folgenden vorgestellten positiven Effekte auf die Rekonvaleszenz tendieren wir bei gegebener Indikation zu einem bevorzugten Einsatz von Antidepressiva aus der Substanzklasse der SSRI bei Patienten mit Schlaganfall. Nachfolgend fokussiert sich aus diesem Grunde diese Übersichtsarbeit auf die Substanzklasse der SSRI. Tabelle 1 gibt einen Überblick über randomisierte kontrollierte Studien, welche die Effektivität von SSRI im Vergleich zu Placebo in der Behandlung einer PSD untersucht haben.
Tab. 1. Zweiarmige, randomisierte, Placebo-kontrollierte Studien über die Wirksamkeit von SSRI zur Behandlung der Post-Schlaganfall-Depression
|
Autoren und Publikationsjahr |
Einschlusskriterien |
Ausschlusskriterien |
Arzneistoff |
Dosis pro Tag |
Dauer der Einnahme |
Aktivgruppe |
Placebo-Gruppe |
Ergebnis |
|
Andersen et al., 1994 |
Ischämischer und hämorrhagischer Schlaganfall Diagnose Depression: HDS >12 |
Depression innerhalb des letzten Jahres Aktuelle Behandlung einer Depression Schwere Demenz Schwere Kommunikationsprobleme Degenerative oder expansive neurologische Erkrankungen Vermindertes Bewusstsein |
Citalopram |
10 mg bei Patienten >66 Jahren 20 mg bei Patienten ≤66 Jahren Dosisverdopplung, wenn innerhalb von 3 Wochen kein Ansprechen auf Behandlung |
6 Wochen |
n=33 (weiblich, n=21) Alter: 68,2±4,2 Jahre Intervall zwischen Schlaganfall und Therapie: 10,6±9,8 Wochen HDS: 19,8±3,2 |
n=33 (weiblich, n=19) Alter: 65,8±9,0 Jahre Intervall zwischen Schlaganfall und Therapie: 13,2±11,0 Wochen HDS: 18,8±2,9 |
HDS nach 6 Wochen: 10,3±4,7 vs. 14,0±4,7 (p<0,005) Unerwünschte Wirkungen: Übelkeit und Erbrechen während der ersten Woche unter Citalopram (p<0,05) Zunahme Asthenie, Abgeschlagenheit oder Müdigkeit während der ersten Woche unter Citalopram (p<0,05) |
|
Frühwald et al., 2003 |
Ischämischer und hämorrhagischer Schlaganfall Diagnose Depression: psychiatrisches Gespräch, mäßig bis schwer depressive Patienten (HDS >15) |
MMSE <20 Mehr als nur milde Kommunikationsschwierigkeiten Relevante Erkrankungen des ZNS, vorherige degenerative oder expansive neurologische Erkrankungen |
Fluoxetin |
20 mg Nach einem Monat: |
12 Wochen |
n=26 (weiblich, n=14)
Intervall zwischen Schlaganfall und Therapie: 11,0±3,9 Tage HDS: 32,8±12,7 BDI: 12,2±5,6 |
n=24 (weiblich, n=7) Alter: 64,0±14,3 Jahre Intervall zwischen Schlaganfall und Therapie: 11,1±3,5 Tage HDS: 30,3±15,0 BDI: 10,9±5,4 |
HDS nach 4 Wochen: 14,7±8,5 vs. 11,7±8,4 (ns) BDI nach 4 Wochen: 6,0±4,5 vs. 5,2±3,5 (ns) |
|
HDS nach 12 Wochen: 9,5±7,9 vs. 11,2±12,4 (ns) BDI nach 12 Wochen: 6,1±5,6 vs. 6,8±7,4 (ns) |
||||||||
|
HDS nach 18 Monaten: 10,8±11,6 vs. 22,2±15,0 (p<0,05) BDI nach 18 Monaten: 5,3±5,6 vs. 8,5±4,9 (p<0,05) |
||||||||
|
Lai et al., 2006 |
Schlaganfall (Unterteilung unbekannt) Depressions HDS >6 |
Unbekannt |
Paroxetin |
20 mg |
8 Wochen |
n=80, 40 vs. 40 (weiblich, n=37) Alter: 60±14 Jahre Weitere Daten liegen nicht vor |
HDS nach 2 Monaten: 12,5±8,4 vs. 21,5±4,3 |
|
|
Murray et al., 2002 |
Ischämischer und hämorrhagischer Schlaganfall Diagnose Depression: psychiatrisches Gespräch, MADRS >9, Unterscheidung major und minor Depression anhand DSM-IV (mit Ausnahme: Zeit-Kriterium von 2 Wochen, medikationsbedingter Ausschluss) Depressive Symptome innerhalb Alter >17 Jahre |
Schwere Beeinträchtigung der Kommunikation Akuter Myokardinfarkt Andere psychiatrische Erkrankungen als Depression Signifikantes Suizidrisiko Antidepressiva Einnahme einen Monat vor Randomisierung Aktuelle Verwendung psychotropischer Medikation oder opioider analgetischer Medikamente Teilnehmer mit weniger als 20% Reduktion in MADRS nach 6 Wochen |
Sertralin |
50 mg |
26 Wochen |
n=62 (weiblich, n=30) Alter: 70,7±9,7 Jahre MADRS [Major Depression (66,1%)]: 20,8±6,2 MADRS [Minor Depression (33,9%)]: 15,2±3,6 Intervall zwischen Schlaganfall und Therapie: 137,3±101,4 Tage |
n=61 (weiblich, n=34) Alter: 70,7±10,1 MADRS [Major Depression (57,4%)]: 22,6±6,2 MADRS [Minor Depression (42,6%)]: 15,5±2,6 Intervall zwischen Schlaganfall und Therapie: 119,0±92,5 Tage |
MADRS nach 6 Wochen: Major Depressive Episode: 12,4±9,6 vs. 13,5±8,1 (ns) Minor Depressive Disorder: 9,1±6,2 vs.10,8±7,4 (ns) |
|
MADRS nach 26 Wochen: Major Depressive Episode: Minor Depressive Disorder: |
||||||||
|
Ponzio et al., 2001 |
Schlaganfall (Unterteilung unbekannt) Diagnose Depression: MADRS >18 Alter 18 bis 85 Jahre MMSE score >23 |
Konkurrierende prädominate psychiatrische Erkrankung Psychotrope Pharmakotherapie Substanz-Abusus/-Abhängigkeit Teilnahme in anderen klinischen Studien Suizidrisiko Konkomitierende Medikation |
Paroxetin |
20 bis 40 mg |
8 Wochen |
n=112 (weiblich, n=52) Alter: 64±11 Jahre Weitere Daten liegen nicht vor |
Anzahl: 117 (weiblich, n=53) Alter: 66±11 Jahre Weitere Daten liegen nicht vor |
Ergebnisse nicht vorliegend |
|
Rampello et al., 2005 |
Kürzlicher (<12 Monate) ischämischer oder hämorrhagischer Schlaganfall Diagnose Depression: psychiatrisches Gespräch, HDS >20, BDI >15 Präsenz einer major oder minor Depression Präsenz einer verzögerten Depression Keine Antidepressiva-Therapie 2 Wochen vor Randomisierung Keine Behandlung mit neuroleptischen Medikamenten, Cinnarizin oder Flunarizin in den letzten 3 Monaten vor Einschluss |
Vorherige degenerative oder expansive neurologische Erkrankungen Psychiatrische Erkrankung (außer Depression) in der Vorgeschichte Schwere Aphasie Schweres kognitives Defizit Chronischer Alkoholismus |
Reboxetin |
8 mg
|
16 Wochen |
n=16 (weiblich, n=9) Alter: 77,5±4 Jahre Intervall zwischen Schlaganfall und Therapie: 12,06±4,23 Wochen HDS: 24,06±1,52 BDI: 20,56±2,16 |
n=15 (weiblich=8) Alter: 77,26±3,6 Jahre Intervall zwischen Schlaganfall und Therapie: 12,26±4,77 Wochen HDS: 24±1,31 BDI: 19,87±1,46 |
HDS nach 4 Wochen: 14,66±1,49 vs. 23,87±1,41 (p<0,01) BDI nach 4 Wochen: 12,56±2,36 vs. 19,8±2,54 (p<0,01) |
|
HDS nach 8 Wochen: 11,8±2 vs. 23,87±2,06 (p<0,01) BDI nach 8 Wochen: 9,62±2,87 vs. 19,47±2,47 (p<0,01) |
||||||||
|
HDS nach 16 Wochen: 9,26±2,15 vs. 22,73±2,4 (p<0,01) BDI nach 16 Wochen: 8,06±3,43 vs. 18,4±3,33 (p<0,01) |
||||||||
|
Wiart et al., 2000 |
Kürzlicher (<3 Monate) ischämischer Schlaganfall Diagnose Depression: psychiatrisches Gespräch, MADRS >19 Stoppen aller antidepressiven oder neuroleptischen Medikation 10 Tage vor Einschluss |
Schwere psychiatrische Probleme, welcher einer Hospitalisation bedürfen Schwere kognitive Beeinträchtigungen (MMS <23) Aphasie Chronischer Alkoholismus |
Fluoxetin |
20 mg |
6 Wochen |
n=16 (weiblich, n=7) Alter: 66,3±7,1 Jahre Intervall zwischen Schlaganfall und Therapie: 47,1±21,6 Tage MADRS: 28,5±7,7 |
n=15 (weiblich, n=9) Alter: 68,9±11,6 Jahre Intervall zwischen Schlaganfall und Therapie: 47,7±19,9 Tage MADRS: 27,2±63 |
MADRS nach 15 Tagen: 20,9±8 vs. 18±6,3 (ns) |
|
MADRS nach 30 Tagen: 15±8,9 vs.18,6±9,4 (ns) |
||||||||
|
MADRS nach 45 Tagen: 11,8±6,7 vs. 18,7±10,0 (p=0,05) |
||||||||
|
Yang et al., 2002 |
Ischämischer und hämorrhagischer Schlaganfall Depressions-Kriterien: HDS-Score >7 |
Unbekannt |
Paroxetin |
20 mg |
4 Monate |
n=64 (weiblich, n=24) Alter: 64±3 Jahre Weitere Daten liegen nicht vor |
Anzahl: 57 (weiblich, n=25) Alter: 63±5 Jahre Weitere Daten liegen nicht vor |
Ergebnisse nicht vorliegend |
Selektive Serotonin-Wiederaufnahmehemmer in der Behandlung nach Schlaganfall
Wirkungsmechanismus
Die SSRI inhibieren die Wiederaufnahme des Neurotransmitters Serotonin in präsynaptische Neurone. Dies führt zu einer Erhöhung der intrasynaptischen Serotoninkonzentration, die über einen nicht vollständig ergründeten Mechanismus zur Entfaltung eines antidepressiven Effekts nach mehrwöchiger Latenz führt [50]. Im Gegensatz zu trizyklischen Antidepressiva weisen SSRI nur eine sehr geringe Wirkung auf Adrenozeptoren, Histamin-Rezeptoren und Muskarin-Rezeptoren auf und sind daher durch eine geringere Häufigkeit von Nebenwirkungen, die das histaminerge System und das autonome Nervensystem betreffen, charakterisiert.
Selektive Serotonin-Wiederaufnahmehemmer in der Behandlung einer Depression
In den vergangenen Jahrzehnten wurde eine Vielzahl an Medikamenten zur Behandlung der Depression eingeführt. In einer umfangreichen Metaanalyse von 117 randomisierten kontrollierten Studien wurden 12 Antidepressiva der neueren Generation hinsichtlich Wirksamkeit und Akzeptanz in der Akutbehandlung der Depression verglichen [11]. Die Substanzen Mirtazapin, Escitalopram, Venlafaxin und Sertralin waren signifikant wirksamer als Duloxetin, Fluoxetin, Fluvoxamin, Paroxetin und Reboxetin. Wichtigstes Ergebnis der vorliegenden Metaanalyse war zudem, dass die SSRI Escitalopram und Sertralin die bevorzugte Wahl bei Erwachsenen mit einer Depression sein sollten, da diese beiden Substanzen die günstigste Relation zwischen Wirksamkeit und Akzeptanz aufwiesen. Eine folgende Metaanalyse der Cochrane Collaboration wies zudem auf eine klinische bedeutsame Überlegenheit von Sertralin gegenüber Fluoxetin in der Behandlung der Depression hin [30].
Positiver Effekt auf das motorische Outcome nach akutem ischämischem Schlaganfall
Neben ihren antidepressiven Eigenschaften sind SSRI deshalb Gegenstand aktueller Diskussion, weil in einer randomisierten kontrollierten Studie (FLAME-Studie) ein positiver Effekt auf das motorische Outcome bei der Behandlung mit Fluoxetin gezeigt wurde [9]. An neun teilnehmenden Studienzentren in Frankreich wurden 118 Patienten nach Schlaganfall mit resultierender Hemiplegie oder Hemiparese (≤55 Punkte im Fugl-Meyer-Test zur Motorik) entweder in den aktiven Fluoxetin-Behandlungsarm (Fluoxetin p.o., 20 mg/Tag) oder in den Placebo-Arm randomisiert. Alle Patienten erhielten zusätzlich Physiotherapie. Der primäre Endpunkt der Studie, die Erholung des motorischen Defizits, wurde mithilfe des Fugl-Meyer-Tests zur Motorik erfasst und war nach Abschluss der 90-tägigen Behandlung in der Fluoxetin-Gruppe signifikant verbessert im Vergleich zu Placebo. Zwischenzeitlich konnten ähnliche Effekte von Fluoxetin in einer randomisierten kontrollierten Studie beobachtet werden, in der 374 Patienten mit ischämischem Schlaganfall entweder einer Behandlung mit Fluoxetin 20 mg/Tag oder Placebo zugeordnet wurden [20]. Die Therapiedauer betrug drei Monate. Nach sechs Monaten waren Patienten in der aktiven Gruppe neurologisch geringer betroffen (NIHSS: 1,74±2,50 versus 1,14±1,71 Punkte) und funktionell unabhängiger (Barthel Index: 87,59±15,72 versus 91,47±12,37) als Patienten im Placebo-Arm. Weitere kleinere prospektive Studien bestätigten einen positiven Einfluss der Behandlung mit einem SSRI auf funktionelle und motorische Outcome-Parameter nach ischämischem Schlaganfall und deuteten darauf hin, dass auch andere SSRI, zum Beispiel Citalopram, einen günstigen Einfluss auf die funktionelle und motorische Erholung nach Schlaganfall haben [1, 53]. In Hinblick auf die in der FLAME-Studie beobachtete Verbesserung des motorischen Outcomes nach Schlaganfall ist ein SSRI-bezogener Klasseneffekt somit wahrscheinlicher als ein substanzspezifischer pharmakodynamischer Mechanismus von Fluoxetin. In einer Metaanalyse der Cochrane Collaboration, in welche insgesamt 4059 Schlaganfallpatienten innerhalb von 12 Monaten nach dem Indexereignis aus 52 Studien eingeschlossen wurden, zeigten Subanalysen eine Verbesserung von funktioneller Abhängigkeit, Behinderung und neurologischen Defiziten bei Schlaganfallpatienten, die nach Einsatz der durch den Schlaganfall verursachten Symptome mit einem SSRI behandelt wurden [34].
Momentan rekrutieren mehrere randomisierte kontrollierte Studien Patienten mit ischämischem Schlaganfall, um die Effekte einer mindestens 6-monatigen Therapie mit Citalopram bzw. Fluoxetin auf das motorische Outcome und die funktionelle Abhängigkeit nach einem ischämischen Schlaganfall zu prüfen [24, 38]. Während der Schwerpunkt bisheriger Untersuchungen auf Patienten mit ischämischen Schlaganfall lag, werden gegenwärtig erstmalig auch Patienten mit hämorrhagischem Schlaganfall in einer kleinen randomisierten kontrollierten Phase-II-Studie zu dem Effekt von Fluoextin 20 mg/Tag auf das neurologische und funktionelle Outcome untersucht [31].
Eine aus tierexperimentellen Studien generierte mechanistische Hypothese in Bezug auf den günstigen Effekt einer Behandlung mit einem SSRI auf das Outcome nach ischämischem Schlaganfall, der unter anderem in der FLAME-Studie beobachtet wurde, basiert vor allem auf der Stimulation der adulten Neurogenese [46]. Daneben wird ein auf tierexperimentellen Studien basierender antiinflammatorischer Mechanismus diskutiert. Insbesondere zeigte sich hierbei ein günstiger Effekt der SSRI-Behandlung auf die Infarktausdehnung und die klinisch neurologischen Defizite in der späten inflammatorischen Post-Akutphase nach Schlaganfall. Die Abbildung 1 skizziert auf tierexperimentellen Daten basierende mechanistische Überlegungen zu SSRI-vermittelten Effekten nach einem ischämischen Schlaganfall. Inwieweit diese Beobachtungen auf den Menschen anwendbar sind, ist jedoch unklar.
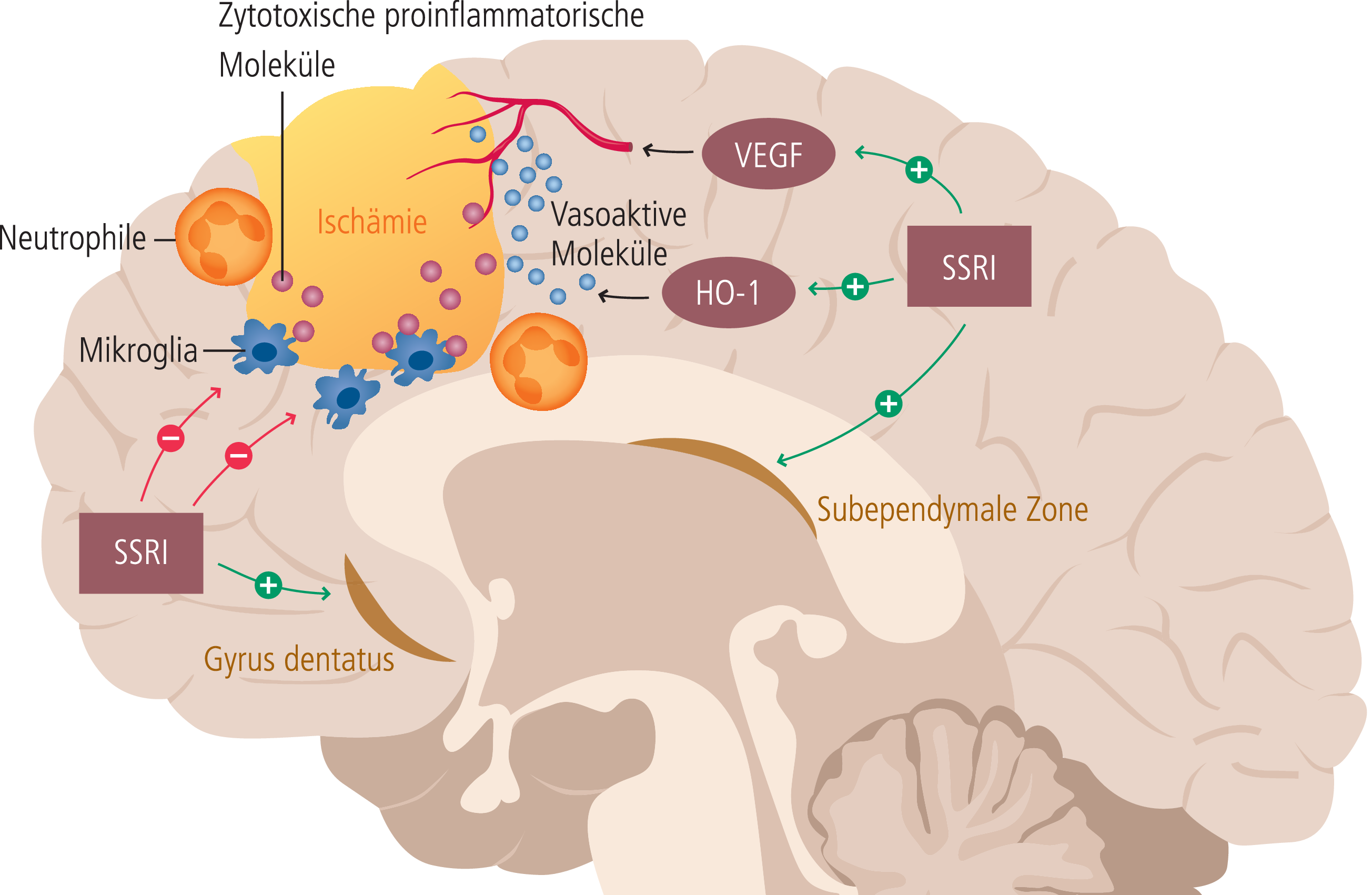
Abb. 1. Auf tierexperimentellen Daten basierende Überlegungen, über welche Mechanismen SSRI einen positiven Effekt auf das Ausmaß einer Ischämie-assoziierten Hirnschädigung haben könnten. 1. Stimulation der Neurogenese in der subependymalen Zone und im hippocampalen Gyrus dentatus; 2. Inhibition der durch Mikroglia und Neutrophilen induzierten Entzündung, vermittelt durch zytotoxische inflammatorische Moleküle; 3. Verbesserung der zerebrovaskulären Autoregulation (HO-1: Hämooxygenase-1; VEGF: Vascular endothelial growth factor) (mod. nach [46])
Unerwünschte Arzneimittelwirkungen und Sicherheitsaspekte in der Behandlung der Post-Schlaganfall-Depression
Generell sind SSRI im Vergleich zu Antidepressiva der früheren Generationen aufgrund ihrer Selektivität hinsichtlich der beteiligten Neurotransmittersysteme mit weniger unerwünschten Wirkungen assoziiert, wodurch die Compliance vorteilhafter zu sein scheint. Gastrointestinale Beschwerden wie Übelkeit, Erbrechen und Diarrhö zählen dabei zu häufigeren Nebenwirkungen der SSRI. Daneben werden nicht selten Unruhe und Schlafstörungen sowie Elektrolytverschiebungen im Sinne einer Hyponatriämie beobachtet, Letztere nicht selten bei kombinierter Gabe mit einem Diuretikum. Zu kardialen Reizleitungsstörungen kann es unter der Einnahme eines SSRI ebenso kommen, weshalb bei älteren Patienten Dosisbeschränkungen für Escitalopram und Citalopram zu beachten sind.
Patienten mit akutem Schlaganfall leiden häufig an vaskulären und metabolischen Begleiterkrankungen wie Diabetes mellitus, arterieller Hypertonie und Dyslipoproteinämie und werden daher zumeist bereits mit multiplen Arzneistoffen behandelt, sodass eine SSRI-Therapie nach Schlaganfall sorgfältig auf etwaige pharmakologische Interaktionen geprüft werden muss. Die Sekundärprophylaxe des ischämischen Schlaganfalls beinhaltet stets die Behandlung mit Thrombozytenfunktionshemmern beziehungsweise bei Vorliegen eines Vorhofflimmerns mit oralen Antikoagulanzien, wobei antikoagulatorische Effekte durch die additive Einnahme von pharmakologischen Wirkstoffen vermindert oder verstärkt werden können. Die Wirkstoffe aus der Substanzklasse der SSRI besitzen unterschiedliches Potenzial für unerwünschte Wirkungen und Interaktionen, sodass stets bei Verordnung die jeweilige Fachinformation zu beachten ist.
Während zahlreiche Studien zu unerwünschten Wirkungen von SSRI in Nicht-Schlaganfallpopulationen vorliegen, existieren kaum spezifische Untersuchungen bei Patienten mit stattgehabtem Schlaganfall [37]. Die größte Akkumulation von Daten zu unerwünschten Wirkungen findet sich in der oben genannten Metaanalyse der Cochrane Collaboration zum Einfluss von SSRI auf das neurologische Outcome nach Schlaganfall [34]. In einer Subanalyse von sieben Studien (n=744) traten epileptische Anfälle im Vergleich zu Kontrollpatienten 2,7-fach häufiger bei SSRI-behandelten Patienten auf. Eine im Jahr 2017 publizierte populationsbasierte Kohortenstudie mit 4688 Schlaganfallpatienten bestätigte diese Beobachtung [10]. Unter einer Behandlung mit einem SSRI war das Risiko für die Ausbildung einer Epilepsie oder epileptischer Anfälle im Vergleich zu Kontrollpatienten 2,5-fach erhöht. Zu erwähnen ist jedoch, dass der Einschluss in diese retrospektive Untersuchung anhand von administrativen Daten (d.h. ICD-Kodierung) erfolgte, was zu einer erheblichen methodischen Unschärfe hinsichtlich der für die Analyse qualifizierenden Diagnosen führte.
SSRI hemmen nicht nur die Wiederaufnahme von Serotonin in präsynaptische Neuronen, sondern verringern auch die Wiederaufnahme von Serotonin in Thrombozyten durch eine Hemmung des membrangebundenen 5-Hydroxytryptamintransporters [32]. Folglich kann es zur Abnahme stimulierender Effekte von Serotonin auf die Thrombozytenaggregation und Hämostase kommen. Zudem scheint die Serotoninkonzentration im Plasma unter Einnahme von SSRI reduziert zu sein, wodurch es zur Beeinträchtigung der Thrombusarchitektur kommen kann [32]. Daneben liegen Studien vor, die neben einer SSRI-vermittelten Hemmung der ADP-, Kollagen- und Thrombin-induzierten Thrombozytenaggregation auch eine Herabregulation von Oberflächenrezeptoren der Thrombozyten, beispielsweise Glykoprotein IIb/IIIa oder P-Selectin, propagieren [32, 44]. Eine Metaanalyse aus 16 Kohorten- und Fall-Kontroll-Studien aus dem Jahr 2012 weist auf ein um 51% erhöhtes Risiko für intrazerebrale und intrakranielle Blutungen unter der Einnahme von SSRI hin, wobei die analysierten Gesamtpopulationen nicht auf Schlaganfallpatienten begrenzt waren [17]. Da sich unter Berücksichtigung epidemiologischer Daten hochgerechnet auf 10000 mit SSRI behandelte Patienten das Risiko nur um das wahrscheinliche Auftreten einer zusätzlichen intrakraniellen Blutung erhöht, schlussfolgern die Autoren, dass das Blutungsrisiko unter SSRI-Therapie insgesamt gering ist. Randomisierte kontrollierte Studien liegen in diesem Kontext nicht vor. Unklar ist daher weiterhin, inwieweit eine Behandlung mit einem SSRI das Risiko für intrakranielle, aber auch systemische Blutungen nach einem Schlaganfall erhöht [37].
In Bezug auf ein potenzielles Blutungsrisiko ist zudem zu beachten, dass einige SSRI durch Interaktion mit dem Cytochrom-P450-(CYP-)Isoenzymsystem über eine Hemmung von CYP2C19 die Überführung von Clopidogrel in seine aktive und damit thrombozytenfunktionshemmende Form hindern können [7]. Neben Acetylsalicylsäure (ASS) wird Clopidogrel zur Sekundärprophylaxe bei Patienten mit ischämischem Schlaganfall angewandt. In einer Kohortenstudie fand sich zwischen Patienten, die aus jeglicher Indikation Clopidogrel und zusätzlich entweder ein das CYP2C19-Enzym stark inhibierendes SSRI (Fluoxetin oder Fluvoxamin; n=9281) oder ein weniger bzw. nichtinhibierendes SSRI (Citalopram, Escitalopram, Paroxetin oder Sertralin; n=44278) verschrieben bekamen, kein Unterschied hinsichtlich gastrointestinaler und intrakranieller Blutungen (Hazard-Ratio [HR] 0,76; 95%-KI 0,5–1,17) [7]. Das Risiko ischämischer Schlaganfälle war unter Einnahme eines CYP2C19-inhibierenden SSRI im Vergleich zu einem nichtinhibierenden SSRI gering erhöht (HR 1,12; 95%-KI 1,01–1,24). Die Studie wurde jedoch an einer nichtselektierten Patientenpopulation durchgeführt, sodass die Generalisierbarkeit auf Schlaganfallpatienten eingeschränkt ist. Eine Kohortenstudie mit 16770 Schlaganfallpatienten bestätigte dennoch unter der Einnahme von SSRI ein um 31% erhöhtes Rezidivrisiko für ischämische Schlaganfälle, das Risiko für hämorrhagische Schlaganfälle war nicht erhöht [24]. Es erfolgte keine Subanalyse nach Art der verordneten SSRI oder zusätzlich eingenommener Thrombozytenfunktionshemmer sowie der Einnahmedauer. Bei Patienten mit Myokardinfarkt, die zusätzlich zu einer Monotherapie mit ASS oder Dualtherapie mit ASS und Clopidogrel ein SSRI erhielten, fand sich im Vergleich zu alleiniger Therapie mit ASS ein 1,4- bzw. 2,35-fach erhöhtes Risiko für systemische und intrakranielle Blutungen [28]. Auch hier handelte es sich allerdings um eine retrospektive Beobachtungsstudie unter Verwendung administrativer Daten mit entsprechenden methodischen Einschränkungen.
Zwei in der Cochrane-Metaanalyse inkludierte Studien berichteten über das Auftreten von Blutungen unter der Behandlung mit Paroxetin beziehungsweise Escitalopram bei Patienten mit Schlaganfall, wobei während des Beobachtungszeitraums von 2 bzw. 12 Monaten das Risiko um 63% im Vergleich zu Placebo erhöht war [34]. In den beiden oben genannten randomisierten kontrollierten Studien zu Fluoxetin fand sich kein erhöhtes zerebrales Blutungsrisiko im Vergleich zu Placebo [9, 20].
Gastrointestinale Nebenwirkungen wurden in der Cochrane-Metaanalyse im Vergleich zu Kontrollpatienten 1,9-fach häufiger bei SSRI-behandelten Patienten beobachtet, wobei hierfür 14 Studien (n=902) berücksichtigt wurden [34].
Abbildung 2 bietet einen Überblick über mögliche Arzneimittelinteraktionen und additive unerwünschte Wirkungen bei gleichzeitiger Einnahme von SSRI und häufig in der Sekundärprophylaxe verwendeten Arzneimitteln (exemplarisch für Sertralin und Fluoxetin).

Abb. 2. Mögliche Arzneimittelinteraktionen zwischen SSRI und Arzneimitteln in der Sekundärprophylaxe des Schlaganfalls (Auswahl). Die Farbtiefe der Felder mit den CYP-Isoenzymen reflektiert deren Wichtung in der Metabolisierung
Evidenzgrad: Expertenmeinung
Für den klinisch tätigen Arzt stellt sich die Frage, ob Patienten mit einem Schlaganfall a priori mit einem Antidepressivum behandelt werden sollten und ob dies dann ein SSRI sein sollte. Generell kann bei allen Patienten eine (indikationsgerechte) Behandlung erwogen werden, bei denen sich klinische Hinweise auf das Vorliegen einer depressiven Störung ergeben. Die Diagnose sollte, je nach Krankheitsphase und Möglichkeit, anhand psychometrischer Fragebögen und psychiatrischer Exploration untermauert werden. Die Erhebung eines ausführlichen psychopathologischen Befunds vor Beginn einer SSRI-Therapie bei einem Patienten mit PSD ist deshalb wichtig, weil sich bereits wenige Tage nach Therapiebeginn eine antriebssteigernde Wirkung entfalten kann, während die antidepressive zumeist erst nach mehreren Wochen einsetzt. Dies ist insbesondere bei Patienten mit aktiver Suizidalität zu berücksichtigen. Im Zweifel sollte stets eine psychiatrische Konsultation erfolgen.
Unabhängig von vorliegenden Studien, die auf einen Benefit einer antidepressiven Therapie bei Schlaganfallpatienten hinweisen, halten wir den prinzipiell erbrachten Wirksamkeitsnachweis von Antidepressiva in der Behandlung einer Depression für hinreichend, um eine Therapie bei Schlaganfallpatienten mit Depression zu rechtfertigen. Bei der Auswahl des Antidepressivums tendieren wir zu einer Bevorzugung von Präparaten aus der Substanzklasse der SSRI, begründet durch deren günstige Wirkungs-/Nebenwirkungsprofile sowie der bei Patienten mit ischämischem Schlaganfall propagierten pleiotropen Effekte.
Unter Würdigung der genannten Studienergebnisse stellt sich zudem die Frage, ob Patienten mit ischämischem Schlaganfall mit motorischen Defiziten Fluoxetin erhalten sollen. Zum einen besteht aufgrund einer resultierenden Behinderung ein hohes Risiko für die Ausbildung einer PSD mit potenziellen nachteiligen Folgen für den rehabilitativen Verlauf, wodurch ein prophylaktischer Einsatz prinzipiell plausibel ist. Eine Risikoabschätzung bezüglich der Ausbildung einer PSD kann anhand der vorgestellten Risikomarker erfolgen. Zum anderen liegen zwei randomisierte kontrollierte Phase-II-Studien vor, die einen positiven Effekt unter der Einnahme von Fluoxetin auf das neurologische Outcome dokumentieren. Sicherheitsbedenken erwuchsen aus diesen Studien nicht. Eine (Off-Label-) Behandlung mit Fluoxetin (ggf. auch mit einem anderen SSRI, da es sich möglicherweise um einen Klasseneffekt handelt) sehen wir daher auf Basis der vorliegenden Studienlage für gerechtfertigt, sofern im Vorfeld eine individuelle Nutzen-Risiko-Abwägung und kritische Prüfung hinsichtlich potenzieller Arzneimittelinteraktionen erfolgt ist. Eine nachfolgend erhöhte Pharmakovigilanz ist dabei selbstverständlich. Es bedarf an dieser Stelle randomisierter kontrollierter Studien, um das tatsächliche Blutungsrisiko unter der Einnahme von SSRI einschätzen zu können. Erste Ergebnisse der in diesem Kontext vorgestellten Studien sind in den nächsten zwei Jahren zu erwarten. Bei Patienten mit hämorrhagischem Schlaganfall ist ein Off-Label-Einsatz von SSRI auf Basis der gegenwärtigen Studienlage nicht begründet.
Interessenkonflikterklärung
IS, TS und KB geben an, dass keine Interessenkonflikte bestehen.
HR hat Honorare für Vorträge und die Teilnahme an einem Expertenbeirat sowie Forschungsbeihilfen erhalten von Abbott, AbbVie, Bayer Health Care, BIAL, Boehringer/Ingelheim, Brittania, Cephalon, Desitin, GSK, Lundbeck, Medtronic, Merck-Serono, Novartis, Orion, Pfizer, TEVA, UCB Pharma, Valeant und Zambon.
Literatur
1. Acler M, Robol E, Fiaschi A, Manganotti P. A double blind placebo RCT to investigate the effects of serotonergic modulation on brain excitability and motor recovery in stroke patients. J Neurol 2009;256:1152–8.
2. Aström M, Olsson T, Asplund K. Different linkage of depression to hypercortisolism early versus late after stroke: a 3-year longitudinal study. Stroke 1993;24:52–7.
3. Ayerbe L, Ayis S, Crichton S, Wolfe CD, et al. The long-term outcomes of depression up to 10 years after stroke; the South London Stroke Register. J Neurol Neurosurg Psychiatry 2014;85:514–21.
4. Barlinn K, Kepplinger J, Puetz V, Illigens BM, et al. Exploring the risk-factor association between depression and incident stroke: a systematic review and meta-analysis. Neuropsychiatric Disease and Treatment 2015;11:1–14.
5. Bodechtel U, Barlinn K, Helbig U, Arnold K, et al. The stroke east Saxony pilot project for organized post-stroke care: a case-control study. Brain Behav 2016;6:e00455.
6. Busch MA, Maske UE, Ryl L, Schlack R, et al. Prävalenz von depressiver Symptomatik und diagnostizierter Depression bei Erwachsenen in Deutschland. Bundesgesundheitsbl 2013;56:733–9.
7. Bykov K, Schneeweiss S, Donneyong MM, Dong YH, et al. Impact of an interaction between clopidogrel and selective serotonin reuptake inhibitors. Am J Cardiol 2017;119:651–7.
8. Carota A, Berney A, Aybek S, Iaria G, et al. A prospective study of predictors of post-stroke depression. Neurology 2005;64:428–33.
9. Chollet F, Tardy J, Albucher JF, Thalamas C, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol 2011;10:123–30.
10. Chou CC, Yen DJ, Lin YY, Wang YC, et al. Selective serotonin reuptake inhibitors and poststroke epilepsy: A population-based nationwide study. Mayo Clin Proc 2017;92:193–9.
11. Cipriani A, Furukawa TA, Salanti G, Geddes JR, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009;373:746–58.
12. De Man-van Ginkel JM, Hafsteinsdóttir TB, Lindeman E, Ettema RG, et al. In-hospital risk prediction for post-stroke depression: development and validation of the post-stroke depression prediction scale. Stroke 2013;44:2441–5.
13. DGPPN. S3-Leitlinie/Nationale VersorgungsLeitlinie – Unipolare Depression [Internet]. 2. Auflage, 2015 (Version 4). AWMF-Register-Nr.: nvl-005. URL: www.dgppn.de/_Resources/Persistent/807ca8f0aa15b6b9a33cc8b204f01c542a39d1e4/S3-NVL_LLreport_depression-2aufl-vers4-llr.pdf (Zugriff am 03.09.2017).
14. Dohmen C, Garlip G, Sitzer M, Siebler M, et al. Post-Stroke-Depression. Fortschr Neurol Psychiat 2006;74:257–62.
15. Douven E, Köhler S, Rodriguez MMF, Staals J, et al. Imaging markers of post-stroke depression and apathy: a systematic review and meta-analysis. Neuropsychol Rev 2017; doi: 10.1007/s11065–017–9356–2.
16. Glodzik-Sobanska L, Slowik A, McHugh P, Sobiecka B, et al. Single voxel proton magnetic resonance spectroscopy in post-stroke depression. Psychiatry Res 2006;148:111–20.
17. Hackam DG, Mrkobrada M. Selective serotonin reuptake inhibitors and brain hemorrhage: a meta-analysis. Neurology 2012;79:1862–5.
18. Hackett ML, Anderson CS, House A, Xia J. Interventions for treating depression after stroke. Cochrane Database Syst Rev 2008;(4):CD003437.
19. Hackett ML, Pickles K. Part I: frequency of depression after stroke: an updated systematic review and meta-analysis of observational studies. Int J Stroke 2014;9:1017–25.
20. He YT, Tang BS, Cai ZL, Zeng SL, et al. Effects of fluoxetine on neural functional prognosis after ischemic stroke: A randomized controlled study in China. J Stroke Cerebrovasc Dis 2016;25:761–70.
21. Herrmann LL, Le Masurier M, Ebmeier KP. White matter hyperintensities in late life depression: a systematic review. J Neurol Neurosurg Psychiatry 2008;79:619–24.
22. Heuschmann PU, Busse O, Wagner M, Endres M, et al. Schlaganfallhäufigkeit und Versorgung von Schlaganfallpatienten in Deutschland. Akt Neurol 2010;37:333–40.
23. Hicks AU, Hewlett K, Windle V, Chernenko G, et al. Enriched environment enhances transplanted subventricular zone stem cell migration and functional recovery after stroke. Neuroscience 2007;146:31–40.
24. Juang HT, Chen PC, Chien KL. Using antidepressants and the risk of stroke recurrence: report from a national representative cohort study. BMC Neurol 2015;15:86.
25. Kalayam B, Alexopoulos GS, Musiek FE, Kakuma T, et al. Brainstem evoked response abnormalities in late-life depression with vascular disease. Am J Psychiatry 1997;154:970–5.
26. Kraglund KL, Mortensen JK, Grove EL, Johnsen SP, et al. TALOS: a multicenter, randomized, double-blind, placebo-controlled trial to test the effects of citalopram in patients with acute stroke. Int J Stroke 2015;10:985–7.
27. Kutlubaev MA, Hackett ML. Part II: predictors of depression after stroke and impact of depression on stroke outcome: an updated systematic review of observational studies. Int J Stroke 2014;9:1026–36.
28. Labos C, Dasgupta K, Nedjar H, Turecki G, et al. Risk of bleeding associated with combined use of selective serotonin reuptake inhibitors and antiplatelet therapy following acute myocardial infarction. CMAJ 2011;183:1835–43.
29. Loubinoux I, Kronenberg G, Endres M, Schumann-Bard P, et al. Post-stroke depression: mechanisms, translation and therapy. J Cell Mol Med 2012;16:1961–9.
30. Magni LR, Purgato M, Gastaldon C, Papola D, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database Syst Rev 2013;7:CD004185.
31. Marquez-Romero JM, Arauz A, Ruiz-Sandoval JL, Cruz-Estrada Ede L, et al. Fluoxetine for motor recovery after acute intracerebral hemorrhage (FMRICH): study protocol for a randomized, double-blind, placebo-controlled, multicenter trial. Trials 2013;14:77.
32. Maurer-Spurej E, Pittendreigh C, Solomons K. The influence of selective serotonin reuptake inhibitors on human platelet serotonin. Thromb Haemost 2004;91:119–28.
33. McCann SK, Irvine C, Mead GE, Sena ES, et al. Efficacy of antidepressants in animal models of ischemic stroke. Stroke 2014;45:3055–63.
34. Mead GE, Hsieh CF, Lee R, Kutlubaev MA, et al. Selective serotonin reuptake inhibitors (SSRIs) for stroke recovery. Cochrane Database Syst Rev 2012;11:CD009286.
35. Mead G, Hackett ML, Lundström E, Murray V, et al. The FOCUS, AFFINITY and EFFECTS trials studying the effect(s) of fluoxetine in patients with a recent stroke: a study protocol for three multicentre randomised controlled trials. Trials 2015;16:369.
36. Meader N, Moe-Byrne T, Llewellyn A, Mitchell AJ. Screening for post-stroke major depression: a meta-analysis of diagnostic validity studies. J Neurol Neurosurg Psychiatry 2014;85:198–206.
37. Mortensen JK, Andersen G. Safety of selective serotonin reuptake inhibitor treatment in recovering stroke patients. Expert Opin Drug Saf 2015;14:911–9.
38. Noonan K, Carey LM, Crewther SG. Meta-analyses indicate associations between neuroendocrine activation, deactivation in neurotrophic and neuroimaging markers in depression after stroke. J Stroke Cerebrovasc Dis 2013;22:e124–35.
39. Nys GMS, van Zandvoort MJE, van der Worp HB, de Haan EHF, et al. Early depressive symptoms after stroke: neuropsychological correlates and lesion characteristics. J Neurol Sci 2005;228:27–33.
40. Paolucci S. Advances in antidepressants for treating post-stroke depression. Expert Opin Pharmacother 2017;18:1011–7.
41. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010;9:689–701.
42. Robinson RG, Jorge RE. Post-stroke depression: A review. Am J Psychiatry 2016;173:221–31.
43. Salter KL, Foley NC, Zhu L, Jutai JW, et al. Prevention of post-stroke depression: Does prophylactic pharmacotherapy work? J Stroke Cerebrovasc Dis 2013;22:1243–51.
44. Serebruany VL, Gurbel PA, O’Connor CM. Platelet inhibition by sertraline and N-desmethylsertraline: a possible missing link between depression, coronary events, and mortality benefits of selective serotonin reuptake inhibitors. Pharmacol Res 2001;43:453–62.
45. Sibolt G, Curtze S, Melkas S, Pohjasvaara T, et al. Post-stroke depression and depression-executive dysfunction syndrome are associated with recurrence of ischaemic stroke. Cerebrovasc Dis 2013;36:336–43.
46. Siepmann T, Penzlin AI, Kepplinger J, Illigens BMW, et al. Selective serotonin reuptake inhibitors to improve outcome in acute ischemic stroke: possible mechanisms and clinical evidence. Brain and Behavior 2015;5:1–7.
47. Sneed JR, Culang-Reinlieb ME. The vascular depression hypothesis: an update. Am J Geriatr Psychiatry 2011;19:99–103.
48. Sun Y, Liang Y, Jiao Y, et al. Comparative efficacy and acceptability of antidepressant treatment in post-stroke depression: a multiple-treatments meta-analysis. BMJ Open 2017;7:e016499.
49. Towfighi A, Ovbiagele B, El Husseini N, Hackett ML, et al. Post-stroke depression: A scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2017;48:e30–43.
50. Vaswani M, Linda FK, Ramesh S. Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Prog Neuropsychopharmacol Biol Psychiatr 2003;27:85–102.
51. Wei N, Yong W, Li X, Zhou Y, et al. Post-stroke depression and lesion location: a systematic review. J Neurol 2015;262:81–90.
52. Weinrieb RM, Auriacombe M, Lynch KG, Lewis JD. Selective serotonin re-uptake inhibitors and the risk of bleeding. Expert Opin Drug Saf 2005;4:337–44.
53. Zittel S, Weiller C, Liepert J. Citalopram improves dexterity in chronic stroke patients. Neurorehabil Neural Repair 2008;22:311–14.
Priv.-Doz. Dr. med. Kristian Barlinn, Isabella Stuckart, Dr. med. Timo Siepmann, Prof. Dr. Heinz Reichmann, Klinik und Poliklinik für Neurologie, Universitätsklinikum Carl Gustav Carus Dresden, Fetscherstraße 74, 01307 Dresden, E-Mail: kristian.barlinn@ukdd.de
Selective serotonin reuptake inhibitors in the treatment of post-stroke depression
About one out of three patients suffer from depression during the first five years following acute stroke. Post-stroke depression is associated with increased morbidity and mortality. The etiology, however, is largely unknown, whereby psychosocial aspects and neurobiological factors such as deficiency of cerebral monoamine neurotransmitters, hypothalamic-pituitary-adrenal dysfunction and inhibition of adult neurogenesis are hypothesized. The identification of post-stroke depression is hampered by possible masking of stroke-related symptoms that commonly hinders adequate treatment of the disease. Several randomized controlled trials have shown a beneficial effect of pharmacological antidepressant therapy on remission frequency and severity of depression. A favorable efficacy/side-effect profile of selective serotonin reuptake inhibitors in general justifies their preferred use in patients with stroke. Data from smaller randomized controlled trials and a meta-analysis also indicate a positive effect of these pharmacological agents on neurological and functional outcomes after stroke, independent of coexistence of depression. Clinical trials, however, are currently investigating this effect in larger stroke patient populations. Antidepressant therapy after stroke should be carefully evaluated for possible pharmacological interactions as patients with acute stroke often suffer from vascular and metabolic disease, and are usually treated with multiple drugs.
Key words: stroke, depression, pharmacotherapy, drug interactions
Psychopharmakotherapie 2018; 25(01):9-20