Holger Petri, Bad Wildungen*
Klassische Antikonvulsiva
Bis 1993 waren nur wenige Wirkstoffe in der Behandlung von Epilepsien verfügbar, von denen Carbamazepin, Phenobarbital, Phenytoin und Valproinsäure zurzeit immer noch häufig eingesetzt werden (Gruppe I, nach [4]). Carbamazepin, Phenobarbital (Metabolit von Primidon) und Phenytoin sind starke Induktoren des Cytochrom-P450(CYP)-Isoenzyms 3A4. CYP3A4 metabolisiert 40% aller den Phase-I-Metabolismus durchlaufenden Arzneistoffe [2]. Zu den Substraten von CYP3A4 gehören unter anderem zahlreiche Arzneimittel aus den Wirkstoffgruppen antineoplastische Mittel, Antiinfektiva, Blut-, Herz- und Kreislaufmittel, Immunsuppressiva, Urologika und ZNS-Mittel [2]. CYP3A4-Induktoren erhöhen die hepatische Clearance dieser Substrate („Victim Drugs“). Es droht ein Therapieversagen wegen erniedrigter AUC-Werte (AUC =Area under the curve, Fläche unter der Konzentrations-Zeit-Kurve), wenn CYP3A4 das Hauptenzym des Abbaus ist. Die Arzneitherapie ist das wichtigste Verfahren in der Epilepsiebehandlung. Etwa 60% aller Patienten müssen lebenslang ein antikonvulsives Medikament einnehmen [4]. Bei der Verordnung potenter CYP3A4-induzierender Substanzen besteht somit das Problem, dass Arzneimittel, die „Victim Drugs“ sind, in hohen Dosen verabreicht werden müssen, um wirksam zu werden.
Zudem ist zu bedenken, dass auch der Metabolismus von endogenen Substraten durch die Induktoren beeinträchtigt werden kann. Vitamin D wird schneller zu inaktiven Metaboliten abgebaut [12]. In der weiteren Folge steigen die Parathormonspiegel mit dem Risiko eines sekundären Hyperparathyreoidismus. Klinisch macht sich dieser durch einen gestörten Knochenstoffwechsel mit Neigung zu Spontanfrakturen und Knochenschmerzen bemerkbar. Auch der mögliche Einfluss der Induktoren auf die Biosynthese und den Metabolismus von körpereigenen Steroidhormonen und Cholesterol sollte bei Langzeitanwendung beachtet werden. Verschiedene Untersuchungen deuten auf eine erhöhte Inzidenz sexueller Funktionsstörungen und Hyperlipidämie hin [12].
Carbamazepin, Phenobarbital und Phenytoin induzieren neben CYP3A4 weitere CYP-Isoenzyme (Tab. 1). Hieraus können sich zusätzlich klinisch relevante Wechselwirkungen ergeben.
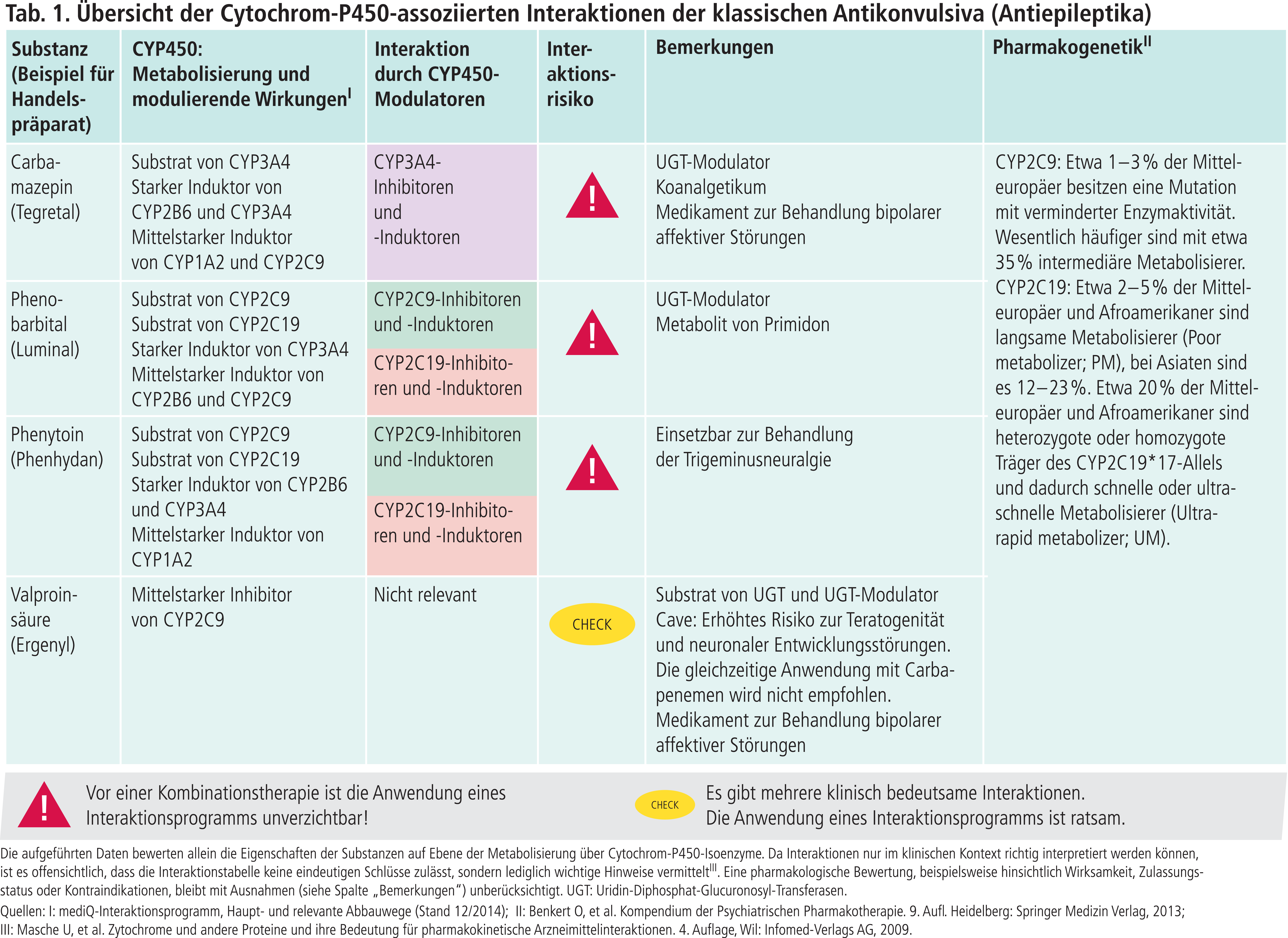
Die Induktoren sind selbst Substrate der CYP-Enzyme. Carbamazepin wird primär über CYP3A4 (Autoinduktion), Phenobarbital über CYP2C19 und Phenytoin über CYP2C9 abgebaut (Tab. 1). Durch Komedikation mit Induktoren (Abb. 1) dieser Isoenzyme sinken die Plasmaspiegel der Antikonvulsiva und es kann zu erneuten Anfällen kommen.
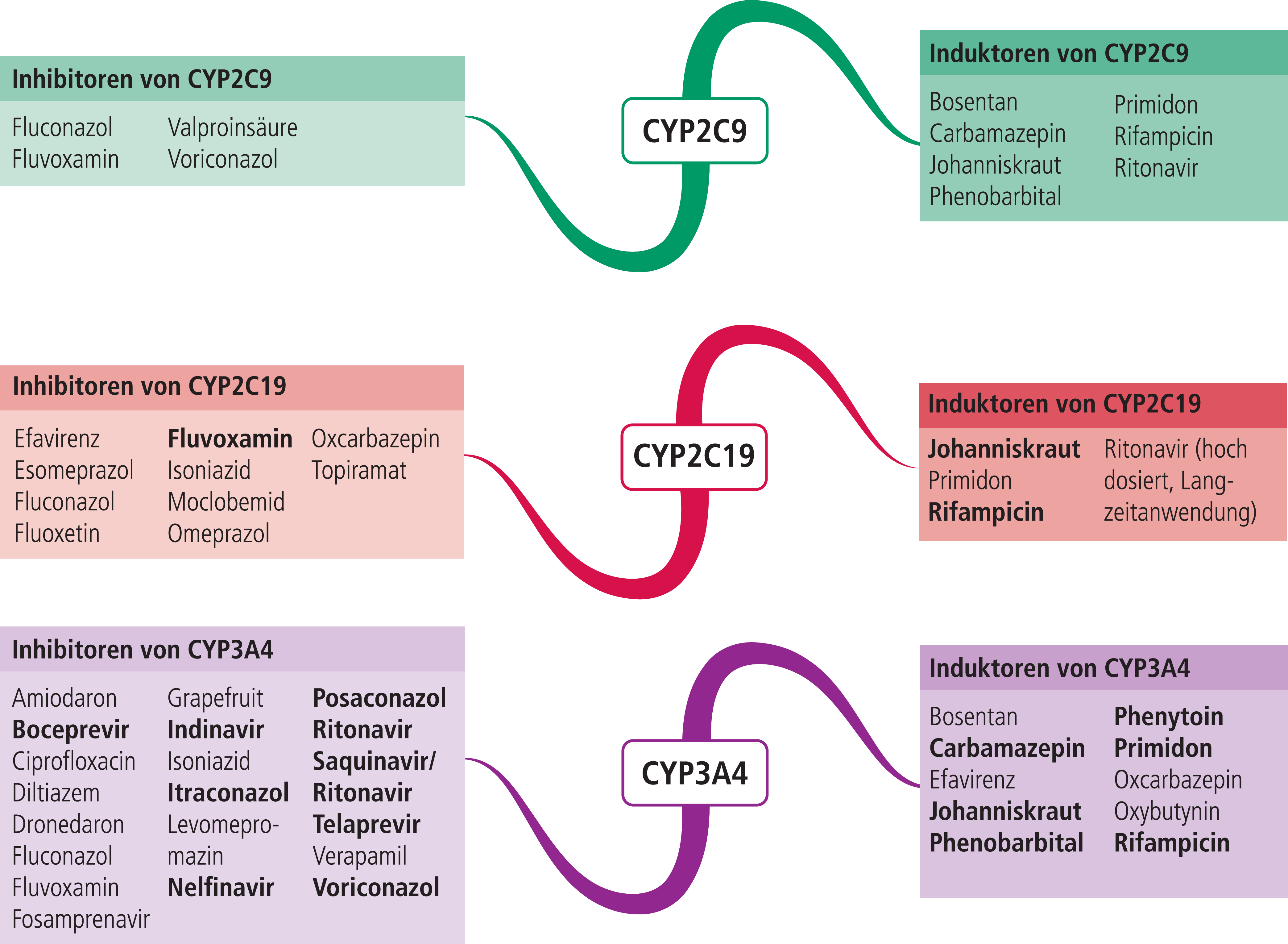
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 2C9, 2C19 und 3A4 (Stand: 12/2014) [Quelle: mediQ-Interaktionsprogramm]
Phenytoin-Intoxikationen sind beschrieben bei gleichzeitiger Anwendung mit potenten CYP2C9-Hemmern [11]. Phenytoin, das zu den wirksamsten Medikamenten zur Durchbrechung eines Status epilepticus gehört, kann schwere Hautreaktionen wie das Stevens-Johnson-Syndrom (SJS) und die toxisch epidermale Nekrolyse (TEN) auslösen. Mutationen im CYP2C9-Gen sind eine mögliche Ursache dieser vermutlich dosisabhängigen Nebenwirkung. Patienten, die Träger des Allels CYP2C9*3 sind und somit eine verminderte Enzymaktivität haben (Poor Metabolizer; PM), sind unter normaler Phenytoin-Dosierung häufiger von schweren Hautreaktionen betroffen als Patienten mit normaler Enzymaktivität (Extensive Metabolizer; EM) [3]. Unter Phenytoin-Therapie wird ein therapeutisches Drug-Monitoring (TDM) dringend empfohlen [9].
Valproinsäure hat ein geringeres pharmakokinetisches Interaktionspotenzial. Klinisch relevante Interaktionen sind mit Substraten von CYP2C9 (nichtsteroidale Antirheumatika, Sulfonylharnstoffe und Vitamin-K-Antagonisten) möglich, da Valproinsäure ein potenter Inhibitor von CYP2C9 ist [10].
Moderne Antikonvulsiva
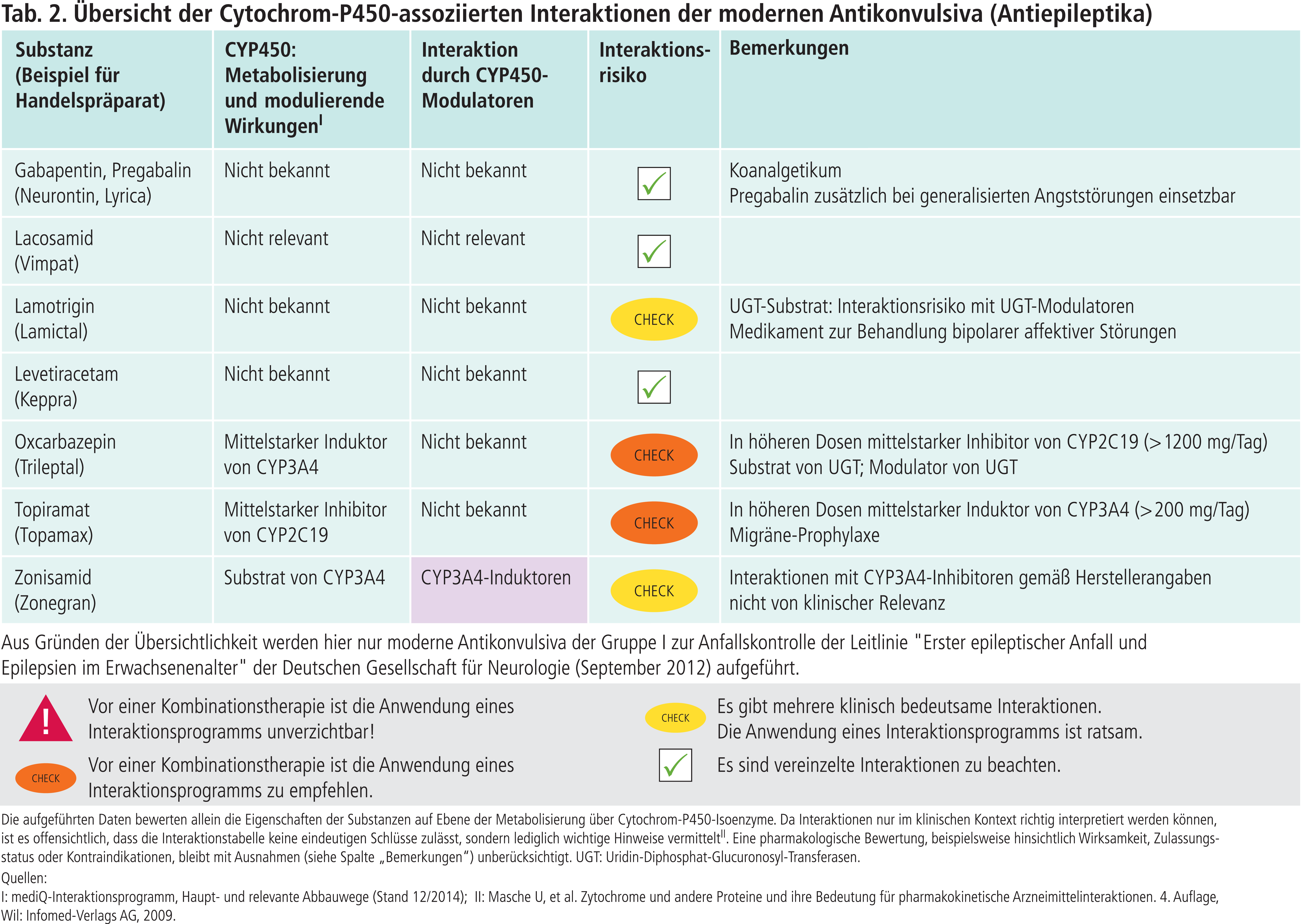
In Gruppe I der Leitlinie der Deutschen Gesellschaft für Neurologie finden sich neben den vier besprochenen klassischen Substanzen acht moderne Antikonvulsiva (Tab. 2). Von diesen treten Gabapentin, Lacosamid, Lamotrigin, Levetiracetam und Pregabalin nicht oder nicht klinisch relevant in Wechselwirkung mit den CYP-Isoenzymen.
Oxcarbazepin und sein pharmakologisch aktiver Metabolit (Monohydroxyderivat, MDH) haben wie Topiramat (>200 mg Tagesdosis) induktive Effekte auf CYP3A4, die im Vergleich zu klassischen Substanzen aber schwächer ausgeprägt sind. Dennoch kann sich die hepatische Clearance des CYP3A4-Substrats Ethinylestradiol in einem Ausmaß erhöhen, dass die kontrazeptive Wirkung nicht sicher gewährleistet ist [1, 7].
Topiramat und Oxcarbazepin (>1200 mg Tagesdosis) sind CYP2C19-Hemmer [6, 7]. Das Antidepressivum Citalopram wird enantioselektiv über CYP2C19 abgebaut mit Bevorzugung des Abbaus von S-Citalopram. Durch potente CYP2C19-Inhibitoren geht diese Stereoselektivität verloren. Die Plasmaspiegel von S-Citalopram steigen, die von R-Citalopram nicht [13]. Dadurch besteht ein erhöhtes Risiko für kardiale Nebenwirkungen.
Zonisamid wird primär über CYP3A4 metabolisiert. Bei gleichzeitiger Anwendung von CYP3A4-Induktoren sollen die Patienten gemäß Fachinformation engmaschig überwacht werden und die Dosierungen nach Bedarf angepasst werden. CYP3A4-Inhibitoren hatten bei gesunden Probanden keine klinisch relevanten Auswirkungen auf die Pharmakokinetik von Zonisamid [8].
Antikonvulsive Kombinationstherapie (Rational Polytherapy)
Wechselwirkungen der antikonvulsiven Kombinationspartner sind nicht nur bei Phase-I-Reaktionen der Biotransformation möglich, sondern können auch bei Phase-II-Reaktionen auftreten. Prominentes Beispiel ist die Interaktion von Lamotrigin mit Valproinsäure. Lamotrigin wird über die UDP-Glucuronosyl-Transferasen (UGT) 1A4 und in geringerem Maß UGT2B7 zu Glucuroniden verstoffwechselt. Valproinsäure hemmt den UGT2B7-abhängigen Abbauweg, sodass ein vorsichtiges Aufdosieren von Lamotrigin notwendig wird. Bei einer Zusatztherapie ohne Valproinsäure mit UGT-Induktoren wie Carbamazepin, Phenobarbital und Phenytoin sind die empfohlenen Tagesdosen von Lamotrigin zur Initial- und Erhaltungstherapie höher [5].
Insbesondere bei Verordnung klassischer Antiepileptika sind die möglichen Interaktionen zahlreich und nur schwer antizipierbar. Ein TDM zur Dosisanpassung und Verlaufskontrolle erhöht die Wahrscheinlichkeit des Ansprechens bei Therapieversagern und verringert das Risiko für Unverträglichkeit oder Vergiftung [9].
Literatur
1. Bialer M, Doose DR, Murthy B, Curtin C, et al. Pharmacokinetic interactions of topiramate. Clin Pharmacokinet 2004;43:763–80.
2. Böhm R, Reinecke K, Haen E, Cascorbi I, et al. Interaktionen mit CYP3A4. Dtsch Apo Ztg 2012;40:58–67.
3. Chung WH, Chang WC, Lee YS, Wu YY, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA 2014;312:525–34.
4. Deutsche Gesellschaft für Neurologie: Erster epileptischer Anfall und Epilepsien im Erwachsenenalter, Stand September 2012.
5. Fachinformation Lamictal®. Stand: August 2014.
6. Fachinformation Topamax®. Stand: Juni 2013.
7. Fachinformation Trileptal®. Stand: Mai 2013.
8. Fachinformation Zonegran®. Stand: Oktober 2013.
9. Hiemke C, Baumann P, Bergemann N, Conca A, et al. AGNP-Konsensus-Leitlinien für therapeutisches Drug-Monitoring in der Psychiatrie: Update 2011. Psychopharmakotherapie 2012;19:91–122.
10. Kämmerer W. Portrait eines Enzyms – CYP2C9. Arzneimitteltherapie 2012;30:123–5.
11. Mamiya K, Kojima K, Yukawa E, Higuchi S, et al. Phenytoin intoxication induced by fluvoxamine. Ther Drug Monit 2001;23:75–7.
12. Mintzer S, Mattson RT. Should enzyme-inducing antiepileptic drugs be considered first-line agents? Epilepsia 2009;50(Suppl 8):42–50.
13. Rocha A, Coelho EB, Sampaio SA, Lanchote VL. Omeprazole preferentially inhibits the metabolism of (+)-(S)-citalopram in healthy volunteers. Br J Clin Pharmacol 2010;70:43–51.
*Nachdruck aus Krankenhauspharmazie 2015;36:29–33.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, Dr. Herbert Hansal, Bad Wildungen, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2015; 22(01)