Christoph Hiemke, Mainz, und Gabriel Eckermann, Kaufbeuren
Entdeckung von pharmakodynamischen Arzneimittelwechselwirkungen in der Psychiatrie
Arzneimittelkombinationen und Polypharmazie mit fünf oder mehr Arzneimitteln sind häufig und nicht selten auch notwendig in der Pharmakotherapie, um einen möglichst guten Behandlungserfolg zu erzielen. Vorteile einer Kombinationsbehandlung wurden für Psychopharmaka erstmalig 1964 von Talbot evaluiert [38], also nur wenige Jahre nach der Entdeckung des ersten Antipsychotikums Chlorpromazin und nach der Entwicklung weiterer Antipsychotika, damals als Tranquilizer bezeichnet. Es wurde festgestellt, dass die Kombination von zwei Antipsychotika bei hospitalisierten psychotischen Patienten gängige Praxis sei. Die klinische Erfahrung spräche dafür, dass mit einer Kombinationstherapie eine bessere Symptomkontrolle erreicht werde als mit einer Monotherapie. Um dies zu belegen, wurden 77 schwer kranke, chronisch schizophrene Patienten in einer Doppelblind-Studie entweder mit Chlorpromazin (150 mg/Tag) oder Trifluoperazin (5 mg) allein oder mit einer Kombination aus beiden Antipsychotika behandelt. Es gab die Option, die Dosis bei Bedarf nach sechs Monaten zu verdoppeln. Nach achtmonatiger Behandlung wurden die drei Gruppen bezüglich Besserung und Nebenwirkungen beurteilt und verglichen. Alle Patienten mit kombinierter Behandlung hatten sich zufriedenstellend gebessert, Patienten mit Chlorpromazin- oder Trifluoperazin-Monotherapie nur zu 72 bzw. 44%. Somit konnte die Überlegenheit einer Kombinationsbehandlung im Vergleich mit Monotherapien erstmalig nachgewiesen werden. Auch bezüglich Verträglichkeit war die Kombination der alleinigen Behandlung mit Trifluoperazin überlegen. Unter der Kombination traten vor allem weniger extrapyramidal-motorische Störungen auf als unter Monotherapie. Dass eine Kombinationsbehandlung in der Psychopharmakotherapie bei vielen Patienten und Störungen nützlich zu sein scheint, bildet sich bis heute im Verordnungsalltag der Psychopharmakotherapie ab. Denn die meisten psychiatrischen Patienten erhalten mehrere Arzneimittel. In einer Analyse an schizophrenen und depressiven Patienten wurde beispielsweise gefunden, dass Patienten mit der Diagnose einer Schizophrenie im Mittel drei Arzneimittel und Patienten mit einer Depression vier Arzneimittel täglich verordnet werden, wobei bei Frauen die Anzahl der Arzneimittel höher ist als bei Männern [20]. Diese Vorgehensweise findet man auch in anderen Ländern, wie den USA [5], den Niederlanden [34] oder Japan [46]. In einer prospektiven naturalistischen Studie mit 796 schizophrenen Patienten erhielten im Verlauf einer einjährigen Beobachtungsphase 58% der Patienten eine antipsychotische Kombinationstherapie, die länger als 60 Tage andauerte [11]. Auch bei der Behandlung von bipolaren Störungen überwiegt die Kombinationstherapie. In einer Untersuchung an 258 ambulanten Patienten [48] erhielten nur 7% eine Monotherapie. Am häufigsten war die Behandlung mit drei Arzneimitteln.
Eine Behandlung mit mehreren Arzneistoffen ist meist unumgänglich, wenn verschiedene Störungen medikamentös behandelt werden müssen. Dies kann im Einzelfall dazu führen, dass mehr als fünf Arzneimittel verordnet werden – mit anderen Worten Polypharmazie. Polypharmazie ist bei Alterspatienten besonders häufig [36]. Bewohner eines Altenheims in Schweden, die zwischen 71 und 100 Jahre alt waren, erhielten vier bis 19 Arzneimittel pro Tag, im Durchschnitt waren es elf Arzneimittel [6]. In einer kanadischen Erhebung erhielt jeder Alterspatient im Mittel sechs Arzneimittel pro Tag [11].
Wechselwirkungsrisiken
Die Behandlung mit mehreren Arzneimitteln bedeutet allerdings fast immer ein erhöhtes Risiko von unerwünschten Arzneimittelwirkungen (UAW). Dies wurde noch vor der ersten Doppelblindstudie von Talbot [38] zur Überlegenheit einer antipsychotischen Kombinationstherapie im Vergleich zu einer Monotherapie erkannt. Es gibt eine Serie von frühen Fallberichten über Intoxikationen, teils schwerwiegend bis hin zu Todesfällen. Sie dokumentieren die Gefährlichkeit der Kombination von beispielsweise Tranylcypromin und Imipramin [1], Imipramin und Chlorpromazin [29] oder Amitriptylin und Thioridazin [43]. Nicht zuletzt durch Kombinationsbehandlungen wurde erkannt, dass trizyklische Antidepressiva und auch Chlorpromazin Atropin-artige Eigenschaften besitzen, also anticholinerg wirken und sich zu einem toxischen Effekt addieren, was einer pharmakodynamischen Wechselwirkung entspricht.
Entdeckung pharmakokinetischer Wechselwirkungen
Die ersten Entdeckungen pharmakodynamischer Wechselwirkungen der seit 1953 neu verfügbaren Psychopharmaka waren allesamt zufällig. Sie haben mit dazu beigetragen, Mechanismen aufzuklären und Rezeptoren zu finden, über die Psychopharmaka erwünschte und unerwünschte Wirkungen entfalten. So wurde erkannt, dass trizyklische Antidepressiva „Dirty Drugs“ sind. Daraus abgeleitete Erkenntnisse gaben den Anstoß, nach selektiveren Wirkstoffen zu suchen. Die Suche war erfolgreich. 1983 wurde mit Zimelidin der erste selektive Serotonin-Wiederaufnahmehemmer (SSRI) zugelassen [3] und unter dem vielversprechenden Namen „Normud“ (für „Nur Mut“) in den Handel gebracht. Doch schon wenige Monate nach Zulassung musste das Präparat wegen schwerwiegender unerwünschter Arzneimittelwirkungen (Guillain-Barré-Syndrom und andere Symptome) wieder vom Markt genommen werden [10]. Es folgten bald darauf Fluvoxamin und Fluoxetin. Dass diese SSRI beide pharmakokinetisch interaktionsträchtige Arzneistoffe sind, nämlich potente Inhibitoren verschiedener Cytochrom-P450(CYP)-Isoenzyme, war zum Zeitpunkt der Zulassung nicht bekannt. Einen ersten Hinweis gab es für Fluoxetin im Rahmen einer Kombinationsstudie mit Diazepam [32]. Die Untersucher fanden, dass Fluoxetin die Elimination von Diazepam verlangsamt, indem es die N-Demethylierung von Diazepam zu Nordazepam hemmt. Der Effekt wurde aufgrund der Ergebnisse psychomotorischer Tests als klinisch nicht relevant beurteilt [32]. Aber schon bald danach wurde für Fluoxetin und Fluvoxamin unter Kombination mit trizyklischen Antidepressiva erkannt, dass das pharmakokinetische Interaktionspotenzial der beiden SSRI von erheblicher klinischer Relevanz ist [8]. Die Entdeckung dieser pharmakokinetischen Wechselwirkungen war ebenfalls unerwartet. Gefunden wurden sie durch therapeutisches Drug-Monitoring (TDM) der trizyklischen Antidepressiva. Durch TDM wurden viele weitere pharmakokinetische Wechselwirkungen identifiziert. Eigene Untersuchungen haben durch TDM zu Tage gebracht, dass Fluvoxamin den Abbau von Clozapin [22] oder Clomipramin [19], Moclobemid die O-Demethylierung von Dextromethorphan [18] und Melperon die O-Demethylierung von Venlafaxin [14] hemmt oder dass Oxybutynin die N-Demethylierung von Clomipramin induziert [15]. Durch Erkenntnisfortschritte in der Pharmakokinetik und Pharmakogenetik seit 1990 und insbesondere dem immer besseren Verständnis des CYP-Enzymsystems konnten viele pharmakokinetische Wechselwirkungen distinkten Isoenzymen zugeordnet werden. Parallel dazu wurden neue Tests etabliert, beispielsweise exprimierte humane CYP-Enzyme, mit denen das CYP-Profil und das pharmakokinetische Interaktionspotenzial eines Arzneistoffs in vitro evaluiert werden können [16]. Von jedem neu eingeführten Arzneimittel ist heute das Interaktionspotenzial im Augenblick der Zulassung bekannt. Seitens der Food and Drug Administration (FDA) gab es 2012 präzise Vorschläge, wann und wie Interaktionsstudien bei der Entwicklung von Arzneimitteln durchzuführen sind [25]. Zufallsentdeckungen von pharmakokinetischen Arzneimittelwechselwirkungen sind daher kaum noch zu erwarten. Bei Auftreten unerwarteter Symptome ist allerdings immer auch eine Arzneimittelwechselwirkung als Ursache in Erwägung zu ziehen und zur Kontrolle eine Messung der Wirkstoffkonzentrationen im Blut durchzuführen.
Die Entdeckungen der vergangenen Jahrzehnte und die damit verbundenen Erkenntnisfortschritte haben dazu geführt, dass Arzneimittelwechselwirkungen und potenzielle Risiken verstehbar und deshalb weitgehend vorhersehbar geworden sind [27]. Die meisten wechselwirkungsbedingten UAW zählen daher inzwischen zu den vermeidbaren Medikationsfehlern [28, 35].
Erkennen und Vermeiden von unerwünschten Arzneimittelwechselwirkungen
Zur Überprüfung von potenziell kritischen Kombinationstherapien und Polypharmazie stehen Lehrbücher [2], Übersichtsarbeiten, umfangreiche Tabellenwerke [2, 23] und in stetig wachsender Zahl computergestützte Programme [44, 45] zur Verfügung. Sie erfassen das in den letzten Jahrzehnten angehäufte Wissen und liefern viele wichtige Hinweise auf potenzielle Risiken. Sie sind hilfreich und notwendig zur Risikominimierung. In der Regel werden dabei Kombinationspaare beurteilt. Die Informationen sind allerdings nicht immer befriedigend. Bei Interaktionschecks werden häufig Warnsignale generiert, obwohl das klinische Risiko gering ist. Und es kommt vor, dass keine Warnung erscheint, weil die Kombination im Programm nicht erfasst worden ist. Bei Polypharmazie ist oft auch nicht klar, ob alle möglichen Kombinationspaare überdacht oder nachgeschlagen werden müssen, um das Risiko einer Arzneimittelwechselwirkung einzuschätzen. Die Kombinationsvielfalt ist riesig. Die Anzahl möglicher Kombinationen (i) zwischen zwei Arzneistoffen lässt sich mit der Funktion
i=(n2–n)/2
beschreiben, wobei n der Anzahl der kombinierten Wirkstoffe entspricht. Bei einem Patienten, der zehn Arzneistoffe einnimmt, gibt es demnach 45 Arzneistoffpaare, zwischen denen bilateral Interaktionen auftreten können. Nach der Roten Liste sind etwa 2200 Wirkstoffe in Deutschland im Einsatz. Berechnet man alle möglichen Kombinationen, gibt es 2418900 mögliche Zweierkombinationen. Diese Zahl potenziert sich weiter, wenn man mögliche Mehrfachkombinationen mit bedenkt. Daraus wird deutlich, dass nicht jede denkbare Arzneistoffkombination in einem Wechselwirkungsprogramm enthalten sein kann. Interaktionsprogramme wie die Scholz-Datenbank, die über die Apotheken-Umschau kostenlos zur Verfügung steht [24], versuchen das Problem der Kombinationsvielfalt durch informationstechnologische Ansätze zu lösen. Dies führt allerdings bei Polypharmazie oft zu verwirrenden Ergebnissen. Und am Ende gibt die Datenbank der Apotheken-Umschau die dringende Empfehlung „Fragen Sie Ihren Arzt oder Apotheker“, die ihrerseits häufig überfordert sind, Wechselwirkungen zu überschauen. Besser ist es daher, Programme wie mediQ.ch [44] (www.mediq.ch) oder psiacOnline [45] (www.psiac.de) zu verwenden, bei denen jede einzelne Wechselwirkung bezüglich ihrer Validität überprüft ist. Nachteil dieser Datenbanken: Viele mögliche Kombinationen sind nicht erfasst.
Aus oben genannten Gründen ist es bei Kombinationstherapie und Polypharmazie unumgänglich, jede verabreichte Arzneimittelliste bezüglich ihres Interaktionsrisikos individuell zu prüfen. Dazu ist es notwendig, das pharmakodynamische und das pharmakokinetische Profil der kombinierten Arzneistoffe zu betrachten. Es muss allerdings nicht jede denkbare Kombination einer Arzneimitteliste überprüft werden. Die Kunst der individuellen Risikoeinschätzung von potenziellen Wechselwirkungen bei Polypharmazie besteht darin, in der Liste der verordneten Arzneimittel interaktionsträchtige Wirkstoffe zu identifizieren – im Sinne einer Signalerkennung. Wenn man die relevanten wechselwirkungspharmakologischen Mechanismen betrachtet, kann relativ einfach festgestellt werden, welche Arten von Risiken bestehen, ob sie in Kauf genommen werden können oder ob eine Kombinationsbehandlung verändert werden muss.
In diesem Beitrag werden die wichtigsten Interaktionsmechanismen zusammenfassend dargestellt und Listen interaktionsträchtiger Arzneistoffe (pharmakodynamische und pharmakokinetische) einschließlich deren Interaktionseffektstärken wiedergegeben. Damit sollen potenzielle pharmakodynamische und pharmakokinetische Interaktionen im Sinne einer Drug Interaction Burden Quantification erfasst werden. Die erstellten Tabellen sollen mit einem vorgeschlagenen Algorithmus verwendet werden, um zu erwartende Wechselwirkungen und deren klinische Relevanz einzuschätzen, ein Werkzeug, um individualisiert zu prüfen und möglichst sicher kombinieren zu können.
Pharmakodynamische Wechselwirkungen
Eine Arzneimittelwechselwirkung liegt vor, wenn die Wirkung eines Arzneimittels durch die Zugabe eines zweiten Arzneimittels verändert wird. Die Wirkung kann gesteigert oder abgeschwächt sein. Generell wird dabei zwischen pharmakokinetischen und pharmakodynamischen Arzneimittelinteraktionen unterschieden [17].
Bei pharmakodynamischen Wechselwirkungen wirken die kombinierten Arzneistoffe am gleichen Rezeptor, Erfolgsorgan oder Regelkreis. Wenn die Interaktionspartner gleichsinnig angreifen, wird die Wirkung verstärkt, wenn ein Partner agonistisch und der andere antagonistisch wirken, wird der Effekt abgeschwächt oder aufgehoben.
Es wird davon ausgegangen, dass die meisten pharmakodynamischen Interaktionen, bei denen es zu einer Wirkverstärkung kommt, vom Behandler bewusst für die Wirkverbesserung im Sinne einer Augmentation oder Potenzierung eingesetzt werden. Verbreitet sind beispielsweise die Kombination von Antidepressiva und Lithium oder die Kombination von zwei Antipsychotika bei unzureichendem Ansprechen. Typische unerwünschte pharmakodynamische Effekte, mit denen unter Kombination verstärkt gerechnet werden muss, sind in Tabelle 1 dargestellt.
Tab. 1. Typische unerwünschte Arzneimittelwirkungen, die sich bei Kombination von Arzneistoffen im Sinne pharmakodynamischer Wechselwirkungen verstärken oder abschwächen können
|
Unerwünschte Wirkung |
Klinische Symptome |
|
Anticholinerge Effekte |
Akkommodationsstörungen, Mundtrockenheit, Sinustachykardie, Obstipation bis hin zum paralytischen Ileus, Harnretention, Glaukom, kognitive Störungen, Delir, Krampfanfälle |
|
Antidopaminerge Effekte |
Hyperprolaktinämie, sexuelle Funktionsstörungen, extrapyramidale Symptome, Tremor, Akathisie, Unruhe, Stürze, verminderte Fahrtauglichkeit |
|
Serotonerge Stimulation |
Gastrointestinale Störungen, Nausea, Erbrechen, Serotonin-Syndrom (Störungen des zentralen, neuromuskulären und vegetativen Nervensystems mit Anstieg von Herzfrequenz und Blutdruck, Übelkeit, Erbrechen, Durchfall, Unruhe, Muskelzuckungen, gesteigerten Reflexen, Hyperthermie, vermehrtem Schwitzen, Tremor, Kopfschmerzen, Tachypnoe, Mydriasis, Akathisie, Koordinationsstörungen, Halluzinationen, Erregungszuständen, Bewusstseinsstörungen und erhöhtem Anfallsrisiko) |
|
Kardiovaskuläre Störungen |
Orthostatische Hypotension, arterieller Hypertonus, Tachykardie, Bradykardie, QTc-Zeit-Verlängerung, Torsade de pointes, ventrikuläre Arrhythmie, Herzstillstand, Myokarditis, Myokardiopathie |
|
Sedierung |
Müdigkeit, Konzentrationsstörungen, kognitive Störungen, Stürze, verminderte Fahrtauglichkeit |
|
Erkrankungen des Blutes |
Leukozytopenie, Agranulozytose, Thrombozytopenie, Blutungen |
|
Leberfunktionsstörung |
Bauchschmerzen, Verdauungsprobleme, Appetitlosigkeit, Müdigkeit, Fieber, Ikterus, Leberversagen, Anstieg der Leberenzymaktivität im Blut |
|
Niereninsuffizienz |
Müdigkeit, Übelkeit, verminderte Harnausscheidung, Flüssigkeitsansammlungen im Körper, Blutdruckanstieg, rötlich-brauner Urin, schaumiger Urin (durch Eiweiße), Knochenschmerzen, Muskelkrämpfe, Muskelschwäche, Nierenversagen, Creatinin-Anstieg in Blut und Urin |
|
Psychische Störungen |
Depression, Halluzinationen, Demenz, Verwirrtheit, Delir |
Bei einer Kombination von Arzneistoffen, die gleichartige unerwünschte Arzneimittelwirkungen (UAW) verursachen können (siehe Fachinformationen), ist davon auszugehen, dass die Wahrscheinlichkeit des Auftretens und die Intensität der UAW zunehmen
Dass sich anticholinerge, antidopaminerge oder Serotonin-stimulierende Effekte, die bei Psychopharmaka und Nichtpsychopharmaka unterschiedlichster Klassen vorkommen, addieren und möglicherweise zu schwerwiegenden UAW führen, erscheint aus dem pharmakologischen Wirkprinzip plausibel. Wenn die in Tabelle 1 aufgeführte Nebenwirkung selten ist und keinem distinkten pharmakologischen Wirkprinzip zuzuschreiben ist, dann fehlt in der Regel ein Nachweis, dass das Risiko einer UAW bei Kombination steigt. Trotzdem wird bei schwerwiegenden UAW wie Arzneimittel-induzierter Agranulozytose in Fachinformationen auf ein potenziell erhöhtes Risiko bei einer Kombination mit einem ähnlichen Nebenwirkungsprofil hingewiesen. Dies ist aus Sicht der Arzneimittelsicherheit auch sinnvoll und vereinzelt ist das erhöhte Risiko auch belegt, so beispielsweise für das Agranulozytoserisiko von Clozapin. Lahdelma und Mitarbeiter [30] fanden durch retrospektive Analyse in Finnland 138 Patienten, die in der Zeit zwischen 1982 und 2007 eine Agranulozytose entwickelten. Die Untersucher stellten fest, dass 40% aller Patienten und 80% der Agranulozytose-Fälle mit fatalem Ausgang zusätzlich ein weiteres Arzneimittel mit Agranulozytose-Risiko eingenommen hatten.
Anticholinerge Eigenschaften eines Arzneistoffs sind erkennbar an der Affinität zu muskarinischen Acetylcholinrezeptoren, die sich in vitro darstellen lassen, an anticholinerger Aktivität im Serum [7] und an klinischen Symptomen [9]. Bei Arzneistoffen mit hoher anticholinerger Aktivität treten unangenehme Effekte (Mundtrockenheit oder Akkommodationsstörungen) unter therapeutischen Dosen und Plasmakonzentrationen auf, unter hohen Konzentrationen toxische Effekte (Obstipation, Harnretention, Glaukom, kognitive Störungen, Delir, epileptische Anfälle, Sinustachykardie). In einer Untersuchung von Hospizpatienten wurden potenzielle Arzneimittelwechselwirkungen bei 223 Patienten analysiert [12]. Die häufigsten Wechselwirkungs-UAW waren additive anticholinerge vor antidopaminergen und kardiovaskulären Effekten. Polypharmazie war nach multivariater Analyse der wichtigste Prädiktor für Wechselwirkungen. Chew und Mitarbeiter [7] haben bei Alterspatienten 107 gängige Arzneistoffe bezüglich ihrer anticholinergen Aktivität von hoch bis nicht nachweisbar klassifiziert. Diese sind in Tabelle 2 wiedergegeben und mit in Deutschland gängigen Arzneistoffen mit anticholinerger Aktivität ergänzt.
Tab. 2. Anticholinerge Aktivitäten von Psychopharmaka und Nichtpsychopharmaka und ihre bei Kombinationsbehandlungen anzunehmende Wirkstärke
|
Anticholinerge Aktivität |
Psychopharmaka |
Nichtpsychopharmaka |
|
Hoch |
Amitriptylin, Clomipramin, Clozapin, Desipramin, Doxepin, Promethazin, Thioridazin, Trifluoperazin, Trimipramin |
Atropin, Benzatropin, Biperiden, Butylscopolaminiumbromid, Carbinoxamin, Darifenacin, Dimenhydrinat, Fesoterodin, Flavoxat, Glycopyrronium, Hyoscyamin, Hydroxyzin, Ipratropiumbromid, Meclizin, Orphenadrin, Oxybutynin, Pirenzepin, Scopolamin, Solifenacin, Tiotropium, Tolterodin, Trihexyphenidyl, Trospium |
|
Moderat |
Chlorpromazin, Diphenhydramin, Loxapin, Nortriptylin, Olanzapin, Paroxetin, Pimozid |
Amantadin, Cimetidin, Cyclobenzapin, Cyproheptadin, Molindon, Oxcarbazepin, Oxycodon |
|
Schwach |
Citalopram, Escitalopram, Fluoxetin, Lithium, Melperon, Mirtazapin, Quetiapin, Temazepam |
Ranitidin |
|
Minimal |
Duloxetin, Diazepam, Phenytoin, Topiramat |
Amoxicillin, Celecoxib, Cephalexin, Digoxin, Diphenoxylat, Fentanyl, Furosemid, Hydrocodon, Lansoprazol, Levofloxacin, Metformin, Propoxyphen |
|
Nicht nachweisbar |
Agomelatin, Alprazolam, Aripiprazol, Bupropion, Buspiron, Carbamazepin, Gabapentin, Haloperidol, Lamotrigin, Lorazepam, Oxazepam, Perphenazin, Risperidon, Sertralin, Trazodon, Valproinsäure, Venlafaxin, Zaleplon, Ziprasidon, Zolpidem |
Acetaminophen, Acetylsalicylsäure, Amlodipin, Atenolol, Atorvastatin, Baclofen, Bisacodyl, Carbidopa, Cetirizin, Ciprofloxacin, Clopidogrel, Codein, Darbepoetin, Diltiazem, Dipyridamol, Enalapril, Epoetin, Famotidin, Fexofenadin, Glipizid, Hydrochlorothiazid, Ibuprofen, Levodopa, Levothyroxin, Lisinopril, Loperamid, Loratadin, Losartan, Lovastatin, Megestrol, Metoprolol, Morphin, Nifedipin, Nitroglycerin, Omeprazol, Pantoprazol, Pioglitazon, Propranolol, Rabeprazol, Rosiglitazon, Simvastatin, Sulfamethoxazol, Tramadol, Trimethoprim, Valsartan, Warfarin |
Die Klassifizierung erfolgte nach Vorgaben von Chew und Mitarbeitern [7] nach der anticholinergen Aktivität im Serum. Diese Liste wurde ergänzt durch Daten von Lertxundi und Mitarbeitern [33]
Kardiotoxische Nebenwirkungen von Psychopharmaka sind zwar selten, aber gefürchtet. Denn sie können zu plötzlichen Todesfällen führen. Ein solches Risiko wurde vor langer Zeit für trizyklische Antidepressiva, später für atypische Antipsychotika und Haloperidol erkannt. In den letzten Jahren wurde auch für Citalopram und weitere Antidepressiva ein solches Risiko identifiziert. Ein Parameter, mit dem potenziell kardiotoxische Wirkungen erfasst werden können, ist die QTc-Zeit [42]. Eine Verlängerung der QTc-Zeit weist auf kardiotoxisches Potenzial hin. QT-Zeit-Verlängerung kann zu Torsade-de-pointes-Tachykardien bis hin zum Herzstillstand führen [38]. In einer Untersuchung an 6790 psychiatrischen Patienten wurde bei 0,9% eine Arzneimittel-induzierte Verlängerung der QTc-Zeit identifiziert [13]. Signifikant waren die Effekte für Haloperidol, Sertindol, Clozapin, Phenothiazine und Citalopram. In Tabelle 3 sind Psychopharmaka und Nichtpsychopharmaka mit QT-Zeit verlängerndem Potenzial dargestellt und bezüglich ihres Risikos klassifiziert in Anlehnung an die AZCERT-Skala [26] und erweitert nach Wenzel-Seifert und Mitarbeitern [42]. Ein moderates bis hohes Risiko besitzen demnach 25 Psychopharmaka, das heißt etwa 20% der in Deutschland verfügbaren Psychopharmaka. Das kardiotoxische Risiko steigt bei Kombination von Arzneistoffen mit QT-Zeit verlängerndem Effekt [39]. Hinzu kommen weitere Risikofaktoren wie Long-QT-Syndrom, Hypokaliämie, Hypomagnesiämie, akuter Myokardinfarkt, Bradykardie, Sepsis, Einnahme von Schleifendiuretika, hohe Wirkspiegel, hohe Dosis und weibliches Geschlecht [39].
Tab. 3. QT-Zeit-verlängerndes Potenzial und Risiko für Torsade de pointes (TdP) von Psychopharmaka und Nichtpsychopharmaka und ihre bei Kombinationsbehandlungen einzuschätzende Wirkstärke
|
QT-Zeit verlängerndes Potenzial und Risiko für TdP |
Psychopharmaka |
Nichtpsychopharmaka |
|
Hoch |
Amitriptylin, Doxepin, Droperidol, Haloperidol i.v., Levomepromazin, Levomethadon, Methadon, Pimozid, Sertindol, Thioridazin |
Amiodaron, Arsentrioxid, Astemizol, Azithromycin, Bepridil, Bretylium, Chinidin, Chloroquin, Cisaprid, Clarithromycin, Diltiazem, Disopyramid, Dofetilid, Domperidon, Erythromycin, Flecainid, Gallopamil, Halofantrin, Ibutilid, Moxifloxacin, Pentamidin, Probucol, Procain, Sevofluran, Sotalol, Sparfloxacin, Terfenadin, Tiaprid, Vandetanib, Verapamil, Vernakalant |
|
Moderat |
Amisulprid, Citalopram, Clozapin, Doxylamin, Escitalopram, Lithium, Melperon, Mianserin, Nortriptylin, Paliperidon, Promethazin, Quetiapin, Risperidon, Venlafaxin, Ziprasidon |
Alfuzosin, Artenimol, Atazanavir, Dolasetron, Dronedaron, Eribulin, Famotidin, Felbamat, Fingolimod, Foscarnet, Granisetron, Indapamid, Isradipin, Lapatinib, Levofloxacin, Moexipril, Nicardipin, Ofloxacin, Ondansetron, Oxytocin, Pasireotid, Perflutren, Ranolazin, Roxithromycin, Saquinavir, Sunitinib, Tacrolimus, Tamoxifen, Telithromycin, Tizanidin, Tolterodin, Vardenafil, Voriconazol |
|
Gering oder fehlend |
Aripiprazol, Agomelatin, Bupropion, Olanzapin |
Die Klassifizierung erfolgte nach der Skala von AZCERT (University-based Center for Education and Research on Therapeutics to foster the safe use of medicines; www.crediblemeds.org) und für Psychopharmaka zusätzlich nach Wenzel-Seifert und Mitarbeitern [42] und Stöllberger und Mitarbeitern [37]. Arzneistoffe mit hohem Risiko sollten nicht kombiniert werden. Bei Identifizierung von Arzneistoff-Kombinationen mit QT-Zeit-verlängerndem Potenzial bzw. Risiko für Torsade de pointes ist individuell zu überprüfen, ob weitere Risikofaktoren vorliegen: Long-QT-Syndrom, Hypokaliämie, Hypomagnesiämie, akuter Myokardinfarkt, Bradykardie, Sepsis, Einnahme von Schleifendiuretika, hohe Wirkspiegel, hohe Dosis, Erregungszustände, Adipositas, weibliches Geschlecht [39].
Serotonin-stimulierende Effekte von Kombinationsbehandlungen sind ebenso wie die kardiotoxischen Effekte ein Risiko, das potenziell tödlich enden kann, das gefürchtete Serotonin-Syndrom [41]. Es äußert sich in Störungen des zentralen, neuromuskulären und vegetativen Nervensystems mit Anstieg von Herzfrequenz und Blutdruck, Übelkeit, Erbrechen, Durchfall, Unruhe, Muskelzuckungen, gesteigerten Reflexen, Hyperthermie, vermehrtem Schwitzen, Tremor, Kopfschmerzen, Tachypnoe, Mydriasis, Akathisie, Koordinationsstörungen, Halluzinationen, Erregungszuständen, Bewusstseinsstörungen und erhöhtem Anfallrisiko. Es kann unter hohen Konzentrationen von Serotonin-stimulierenden Arzneistoffen in Monotherapie auftreten, kommt jedoch im Alltag fast ausschließlich bei Kombinationstherapien vor. Gut bekannt ist die kontraindizierte Kombination von SSRI und Monaminoxidasehemmern. In einer großen Post-mortem-Analyse in Finnland wurden aus 37367 toxikologisch untersuchten Fällen 267 Fälle identifiziert, bei denen Arzneimittelkombinationen die wahrscheinliche Todesursache waren [31]. 58% dieser Fälle waren auf eine Serotonin-Überstimulation zurückzuführen. In Tabelle 4 sind Serotonin-stimulierende Arzneistoffe zusammengestellt.
Tab. 4. Serotonin-stimulierende Psychopharmaka und Nichtpsychopharmaka, die bei Kombination ein Serotonin-Syndrom auslösen können
|
Psychopharmaka |
Nichtpsychopharmaka |
|
Amitriptylin, Amoxapin, Buspiron, Carbamazepin, Citalopram, Clomipramin, Dapoxetin, Doxepin, Duloxetin, Escitalopram, Fluoxetin, Fluvoxamin, Johanniskraut, Lithium, Metamphetamin, Methadon, Mirtazapin, Moclobemid, Nortriptylin, Paroxetin, Sertralin, Sibutramin, Tranylcypromin, Trazodon, Trimipramin, Tryptophan, Valproinsäure, Venlafaxin, Vortioxetin, Ziprasidon Cocain, LSD, MDMA (Ecstasy) |
Almotriptan, Chlorpheniramin, Cocain, Cyclobenzaprin, Dextromethorphan, Eletriptan, Fentanyl, Frovatriptan, Granisetron, Levomethorphan, Levorphanol, Linezolid, Methylenblau, Naratriptan, Ondansetron, Pentazocin, Pethidin, Phenelzin, Phentermin, Rizatriptan, Selegilin, Sumatriptan, Tapentadol, Tramadol, Zolmitriptan |
Ein Serotonin-Syndrom kann bei hohen Dosen unter Monotherapie auftreten, die meisten Intoxikationen finden bei Kombination von zwei oder mehr Serotonin-stimulierenden Wirkstoffen statt. Für diese Substanzklasse liegen in der Literatur keine Angaben zur graduierten Einschätzung des Serotonin-stimulierenden Potenzials der Einzelsubstanzen vor. Zusammengestellt nach Volpi-Abadie und Mitarbeitern [41].
Pharmakokinetische Wechselwirkungen
Bei pharmakokinetischen Interaktionen kommt es durch einen Wirkstoff A zu einer verlangsamten oder beschleunigten Clearance und damit zu einer Erhöhung oder Erniedrigung der Konzentration eines anderen Wirkstoffs B und damit zu Änderungen der Wirkstärke von B. Pharmakokinetische Interaktionen können prinzipiell in allen pharmakokinetischen Phasen auftreten, das heißt während der Absorption, Distribution, Metabolisierung oder Exkretion der Arzneistoffe. Die meisten Psychopharmaka werden oral eingenommen. Die Absorption der in der Regel lipophilen Wirkstoffe erfolgt großenteils während der Darmpassage.
Von wesentlicher klinischer Bedeutung sind Interaktionen in der Phase der Metabolisierung von Psychopharmaka. Bis auf wenige Ausnahmen müssen die meist lipophilen Psychopharmaka metabolisiert werden, um sie über die Niere in gut wasserlöslicher Form ausscheiden zu können. Dies geschieht in zwei Phasen. In Phase-I-Reaktionen wird eine funktionelle Gruppe, häufig eine Hydroxylgruppe, freigelegt oder eingeführt. In Phase II folgt eine Konjugation an die funktionelle Gruppe, meist eine Glucuronidierung. Für pharmakokinetische Wechselwirkungen am wichtigsten sind die in der Leber durch Enzyme des Cytochrom-P450(CYP)-Systems katalysierten Phase-I-Reaktionen. Durch Hemmung oder Induktion der Enzyme kommt es zu einer veränderten Clearance der Arzneistoffe und damit zu veränderten Wirkspiegeln und veränderter Wirkung. Von den 57 aktiven CYP-Enzymen, die im menschlichen Körper vorkommen können [47], sind sieben für die Metabolisierung von Psychopharmaka bedeutsam: CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 und CYP3A4.
Pharmakokinetische Wechselwirkungen sind in erster Linie immer dann zu erwarten, wenn ein Arzneistoff ein bevorzugtes Substrat eines distinkten CYP-Enzyms und ein Kombinationspartner ein Inhibitor oder Induktor dieses Enzyms ist. Ob es aufgrund des angestiegenen Wirkspiegels zu einem klinisch relevanten Interaktionseffekt kommt, hängt von der Hemmstärke des Inhibitors ab, aber auch von der therapeutischen Breite des Arzneistoffs, dessen Metabolisierung gehemmt wird.
Zahlreiche In-vitro-Studien haben für viele Arzneistoffe inhibitorische Eigenschaften festgestellt. Solche Substanzen wurden in Tabellen als Inhibitoren eingetragen, selbst wenn für die Hemmeffekte supratherapeutische Konzentrationen eingesetzt worden waren. Dies und das Fehlen von In-vivo-Studien hat dazu geführt, dass viele Tabellen, die zum Prüfen auf potenzielle pharmakokinetische Wechselwirkungen benutzt werden, für die Praxis zu viele Inhibitoren enthalten. So werden beispielsweise in einigen Tabellen Haloperidol oder Metoprolol als CYP2D6-Inhibitoren geführt, obwohl diese Eigenschaft nur bei toxischen Konzentrationen bedeutsam ist. Es ist daher wichtig, CYP-Inhibitoren (und auch Induktoren) bezüglich ihres Wechselwirkungspotenzials zu klassifizieren. In einem Positionspapier der FDA wurde eine Klassifizierung auf der Basis pharmakokinetischer Untersuchungen vorgenommen [25]. Als starke Inhibitoren wurden solche Substanzen eingestuft, bei denen die Plasmaspiegel kombinierter Arzneistoffe mindestens fünffach steigen, und als moderat, wenn sie um mehr als das Doppelte steigen. Nach diesem Prinzip klassifizierte CYP-Inhibitoren sind in Tabelle 5 zusammengestellt. Aufgrund der strengen Selektionskriterien werden in der Tabelle 5 nur neun Psychopharmaka aufgeführt, bei denen man im klinischen Einsatz mit einer starken oder moderaten Hemmung rechnen muss.
Tab. 5. Psychopharmaka und Nichtpsychopharmaka, die klinisch relevante Inhibitoren distinkter Cytochrom-P450(CYP)-Enzyme sind
|
CYP-Enzym |
Psychopharmaka |
Nichtpsychopharmaka |
|
CYP1A2 |
Fluvoxamin, Perazin |
Ciprofloxacin, Enoxacin, Methoxsalen, Mexiletin, Phenylpropanolamin, Thiabendazol, Vemurafenib, Zileuton |
|
CYP2B6 |
Keine bekannt |
Keine bekannt |
|
CYP2C9 |
Keine bekannt |
Amiodaron, Fluconazol, Miconazol |
|
CYP2C19 |
Fluvoxamin, Fluoxetin, Moclobemid |
Esomeprazol, Omeprazol, Voriconazol |
|
CYP2D6 |
Bupropion, Duloxetin, Fluoxetin, Melperon, Paroxetin |
Chinidin, Terbinafin |
|
CYP2E1 |
Clomethiazol |
para-Kresol (p-Hydroxytoluol, Naturstoff, vor allem in Teer) |
|
CYP3A4 |
Keine bekannt |
Amprenavir, Aprepitant, Atazanavir, Boceprevir, Ciprofloxacin, Crizotinib, Clarithromycin, Conivaptan, Diltiazem, Erythromycin, Grapefruitsaft, Indinavir, Itraconazol, Ketoconazol, Nelfinavir, Posaconazol, Ritonavir, Saquinavir, Telaprevir, Telithromycin, Verapamil, Voriconazol |
Die Hemmwirkung der aufgeführten Inhibitoren ist so stark, dass bei Kombination mit betroffenen Arzneistoffen, die bevorzugt über das gehemmte Enzym abgebaut werden, die Plasmakonzentrationen der betroffenen Arzneistoffe auf das Doppelte oder höher steigen. Bei jeder Kombinationsbehandlung muss immer eine sorgfältige Prüfung potenzieller pharmakokinetischer Wechselwirkungen vorgenommen werden (siehe Abb. 1). Die Klassifizierung der Wirkstoffe als „klinisch relevante“ Inhibitoren wurde in Anlehnung an einen Vorschlag der FDA vorgenommen, die diese Arzneistoffe auf der Basis pharmakokinetischer Untersuchungen als starke oder moderate Inhibitoren eingestuft [25] hat. Die Liste wurde geringfügig erweitert [4, 21] hat. Wirkstoffe mit gering ausgeprägtem Interaktionspotenzial sind nicht aufgeführt. Sie können bei hohen Dosierungen und Intoxikationen klinisch bedeutsam werden.
Bei Induktion eines Enzyms kommt es zu einer Konzentrationserhöhung des CYP-Enzyms, in der Regel durch Stimulation der Proteinsynthese. Bei einer Behandlung mit Carbamazepin und auch anderen Antikonvulsiva ist seit langem bekannt, dass zwei bis drei Wochen nach Beginn der Behandlung eine Dosissteigerung notwendig ist, da der Arzneistoff seinen eigenen Metabolismus induziert. Der induktive Effekt von Carbamazepin hat auch bei Kombination mit Psychopharmaka eine Bedeutung. Wenn nämlich das kombinierte Psychopharmakon Substrat von CYP3A4 ist, dann sollte mit einem beschleunigten Abbau gerechnet werden.
Analog zur Graduierung von Inhibitoren wurden im Positionspapier der FDA [25] auch Induktoren klassifiziert. Ein Arzneistoff wurde als starker Induktor eingestuft, wenn die Plasmaspiegel kombinierter Arzneistoffe um mindestens 80% sinken, und als moderat, wenn sie um mindestens 50% abfallen. Unter Berücksichtigung dieser Klassifizierung sind in Tabelle 6 Arzneistoffe aufgelistet, die als mindestens moderate Induktoren eingestuft wurden, da dies als klinisch relevant anzusehen ist.
Tab. 6. Psychopharmaka und Nichtpsychopharmaka, die klinisch relevante Induktoren distinkter Cytochrom-P450(CYP)-Enzyme sind
|
CYP-Enzyme |
Psychopharmaka |
Nichtpsychopharmaka |
|
CYP1A2 |
Keine bekannt |
Rauchen (Benzpyrene nicht Nicotin), Montelukast |
|
CYP2B6 |
Keine bekannt |
Efavirenz, Rifampicin |
|
CYP2C9 |
Carbamazepin |
Rifampicin |
|
CYP2C19 |
Einige Ginkgo-Präparate |
Rifampicin |
|
CYP2D6 |
Keine bekannt |
Keine bekannt |
|
CYP2E1 |
Ethanol |
Keine bekannt |
|
CYP3A4 |
Carbamazepin, Johanniskraut (Hyperforin), Modafinil , Phenytoin |
Avasimib, Bosentan, Efavirenz, Etravirin, Rifampicin |
Die induzierende Wirkung der aufgeführten Arzneistoffe ist so stark, dass bei Kombination mit betroffenen Arzneistoffen (Victim Drugs), die bevorzugt über das induzierte Enzym abgebaut werden, die Plasmakonzentrationen der betroffenen Arzneistoffe auf weniger als die Hälfte abfallen, sodass das Risiko von Wirkverlust besteht. Wenn die verordnete Arzneimittelliste einen der oben aufgeführten Induktoren enthält, muss weiter geprüft werden, ob die Arzneimittelliste „Victim Drugs“ enthält (siehe hierzu Tab. 7). Induktoren und Victim Drugs sollten nur dann kombiniert werden, wenn die pharmakokinetische Interaktion therapeutisch genutzt werden soll. Die Klassifizierung der Wirkstoffe als „klinisch relevante“ Induktoren wurde in Anlehnung an einen Vorschlag der FDA vorgenommen, die diese Arzneistoffe auf der Basis pharmakokinetischer Untersuchungen als starke oder moderate Induktoren eingestuft hat [25]. Die Liste wurde geringfügig erweitert [4, 21].
Die hemmende oder induzierende Wirkung der in Tabelle 5 und 6 gelisteten Arzneistoffe spielt bei Kombinationstherapie/Polypharmazie dann eine Rolle, wenn die Liste der eingenommenen Arzneimittel neben einem Inhibitor (Tab. 5) oder Induktor (Tab. 6) einen Arzneistoff enthält, der bevorzugt über das gehemmte oder induzierte Enzym abgebaut wird [4, 21, 25]. Potenziell betroffene Arzneistoffe, die bevorzugt über ein distinktes CYP-Isoenzym abgebaut werden, sind in Tabelle 7 gelistet.
Tab. 7. Psychopharmaka und Nichtpsychopharmaka, die bevorzugte Substrate distinkter Cytochrom-P450(CYP)-Enzyme sind
|
CYP-Enzym |
Psychopharmaka |
Nichtpsychopharmaka |
|
CYP1A2 |
Agomelatin, Chlorpromazin, Clomipramin, Clozapin, Coffein, Cyamemazin, Duloxetin, Imipramin, Levomepromazin, Melatonin, Olanzapin, Ropinirol |
Flutamid, Frovatriptan, Propranolol, Rasagilin, Riluzol, Ropivacain, Theophyllin, Tizanidin, Zolmitriptan |
|
CYP2B6 |
Bupropion, Methadon, Sertralin |
Efavirenz, Selegilin |
|
CYP2C9 |
Phenytoin, Tetrahydrocannabinol, Venlafaxin, Vortioxetin |
Celecoxib, Warfarin |
|
CYP2C19 |
Amitriptylin, Citalopram, Clomipramin, Clozapin, Clobazam, Desvenlafaxin (O-Desmethylvenlafaxin), Diazepam, Doxepin, Escitalopram, Imipramin, Loxapin, Moclobemid, Nordazepam, Perazin, Perphenazin, Promazin, Sertralin, Tetrahydrocannabinol, Trimipramin, Venlafaxin |
Esomeprazol, Lansoprazol, Omeprazol, Selegilin |
|
CYP2D6 |
Amitriptylin, Aripiprazol, Atomoxetin, Chlorpromazin, Chlorprothixen, Clomipramin, Dapoxetin, Desipramin, Diphenhydramin, Donepezil, Flupentixol, Fluphenazin, Fluvoxamin, Galantamin, Haloperidol, Imipramin, Levomepromazin, Nortriptylin, Opipramol, Perphenazin, Promethazin, Risperidon, Sertindol, Thioridazin, Venlafaxin, Vortioxetin, Zotepin, Zuclopenthixol |
Alfentanil, Ajmalin, Alprenolol, Benztropin, Carvedilol, Codein, Darifenacin, Dextromethorphan, Dihydrocodein, Diphenhydramin, Encainid, Flecainid, Hydroxyzin, Indoramin, Metoclopramid, Metoprolol, Mexiletin, Nebivolol, Ondansetron, Penbutolol, Pindolol, Prajmalin, Propafenon, Tamoxifen, Tolterodin |
|
CYP2E1 |
Ethanol, Disulfiram |
Paracetamol |
|
CYP3A4 |
Alprazolam, Aripiprazol, Bromazepam, Bromocriptin, Bromperidol, Brotizolam, Buprenorphin, Carbamazepin, Dapoxetin, Dextromethorphan, Diazepam, Disulfiram, Donepezil, Ethosuximid, Flunitrazepam, Fluoxetin, Galantamin, Haloperidol, Imipramin, Lisurid, Lurasidon, Methadon, Midazolam, Norfluoxetin, Perazin, Pimozid, Prazepam, Quetiapin, Risperidon, Sibutramin, Trazodon, Triazolam, Zaleplon, Ziprasidon, Zolpidem, Zopiclon |
Alfentanil, Aprepitant, Amantadin, Amiodaron, Androsteron, Astemizol, Atorvastatin, Budesonid, Chinidin, Chloroquin, Ciclosporin, Cisaprid, Cyclophosphamid, Cortison, Dapson, Darifenacin, Darunavir, Dasatinib, Dexamethason, Dihydroergotamin, Diltiazem, Doxycyclin, Dronedaron, Eletriptan, Eplerenon, Ergotamin, Erythromycin, Ethinylestradiol, Everolimus, Felodipin, Fentanyl, Fluticason, Indinavir, Lidocain, Loratadin, Lovastatin, Maraviroc, Nateglinid, Nelfinavir, Nifedipin, Nimodipin, Nisoldipin, Omeprazol, Pantoprazol, Propafenon, Ritonavir, Saquinavir, Sildenafil, Sirolimus, Simvastatin, Tacrolimus, Tadalafil, Tamoxifen, Terfenadin, Testosteron, Ticagrelor, Tipranavir, Tolvaptan, Tramadol, Vardenafil, Verapamil |
Bei Kombination mit einem Inhibitor (siehe Tab. 5) ist die Elimination vermindert und es besteht das Risiko einer Intoxikation, bei Kombination mit einem Induktor (Tab. 6) wird die Elimination beschleunigt und es besteht das Risiko von Wirkverlust. Zur Risikominimierung sollten Inhibitoren (Tab. 5) oder Induktoren (Tab. 6) mit Arzneistoffen dieser Liste nur dann kombiniert werden, wenn mit keiner pharmakokinetischen Wechselwirkung zu rechnen ist oder wenn die Wechselwirkung erwünscht ist und therapeutisch genutzt werden soll. Die Einstufung der Wirkstoffe als „bevorzugte Substrate“ erfolgte in Anlehnung an Empfehlungen der FDA [25] und nach den Konsensus-Leitlinien für TDM in der Psychiatrie [21].
Lithium-Interaktionen sind naturgemäß keine metabolischen Interaktionen, aber sie gehören zu den pharmakokinetischen Wechselwirkungen. Durch Zugabe oder Weglassen einer Substanz können die Wirkspiegel von Lithium steigen oder abfallen, in erster Linie durch Veränderung der renalen Exkretion. Dies kann von erheblicher klinischer Relevanz sein. Das häufigste Problem ist die Kombination von Lithium mit nichtsteroidalen Antiphlogistika wie Ibuprofen, Diclofenac oder Indomethacin [2]. Nichtsteroidale Antiphlogistika hemmen die Prostaglandinsynthese. Dies reduziert die Nierendurchblutung und die glomeruläre Filtrationsrate, was zum Anstieg der Lithium-Spiegel führt. Da Lithium eine sehr geringe therapeutische Breite hat, geraten die Lithium-Spiegel sehr rasch in den toxischen Bereich. Gleiches gilt für Cyclooxygenase-2-Hemmer (Coxibe), durch die eine gefährliche Erhöhung des Lithium-Blutspiegels möglich ist. Bei nichtsteroidalen Antiphlogistika besteht für Lithium-Patienten das Problem, dass diese Substanzen frei verkäuflich sind. Somit ist es jederzeit möglich, dass ein Lithium-Patient zu diesen Arzneistoffen ohne ärztliche Kontrolle greift und dann einen schweren Schaden durch eine Lithium-Vergiftung erleiden kann. Deshalb ist es notwendig, jeden Lithium-Patienten vor dieser Selbstmedikation zu warnen.
Auch durch die Gabe von ACE-Hemmern (Angiotensin-Konversionsenzymhemmer) oder Angiotensin-II-Rezeptorantagonisten (Sartane) kann der Lithium-Spiegel steigen, hier ist insbesondere im Alter eine engmaschige Kontrolle des Lithium-Spiegels vonnöten. Schließlich ist noch darauf hinzuweisen, dass in einigen Fällen eine Lithium-Intoxikation bei der Zugabe von Topiramat beobachtet wurde. Topiramat kann die renale Exkretion von Lithium beeinträchtigen. Es ist ein potenter Inhibitor der Carboanhydrase. Durch Carboanhydrasehemmung wird die Reabsorption von Bicarbonat und Natrium reduziert, es kann zu einer Natrium-Depletion und metabolischen Azidose kommen. Natrium-Depletion führt zu einem Anstieg der Lithium-Spiegel [2].
Algorithmus zur Identifizierung klinisch relevanter Wechselwirkungen
Aus dem Rezeptorprofil eines Arzneistoffs lässt sich auf potenzielle pharmakodynamische Wechselwirkungen schließen, aus CYP-Eigenschaften auf pharmakokinetische. Die für diesen Beitrag erstellten Tabellen können genutzt werden, um eine systematische Prüfung potenzieller pharmakodynamischer und pharmakokinetischer Wechselwirkungen vorzunehmen, die mit hoher Wahrscheinlichkeit klinisch relevant sind. Dazu wurde ein Algorithmus entwickelt, der in Abbildung 1 dargestellt ist.
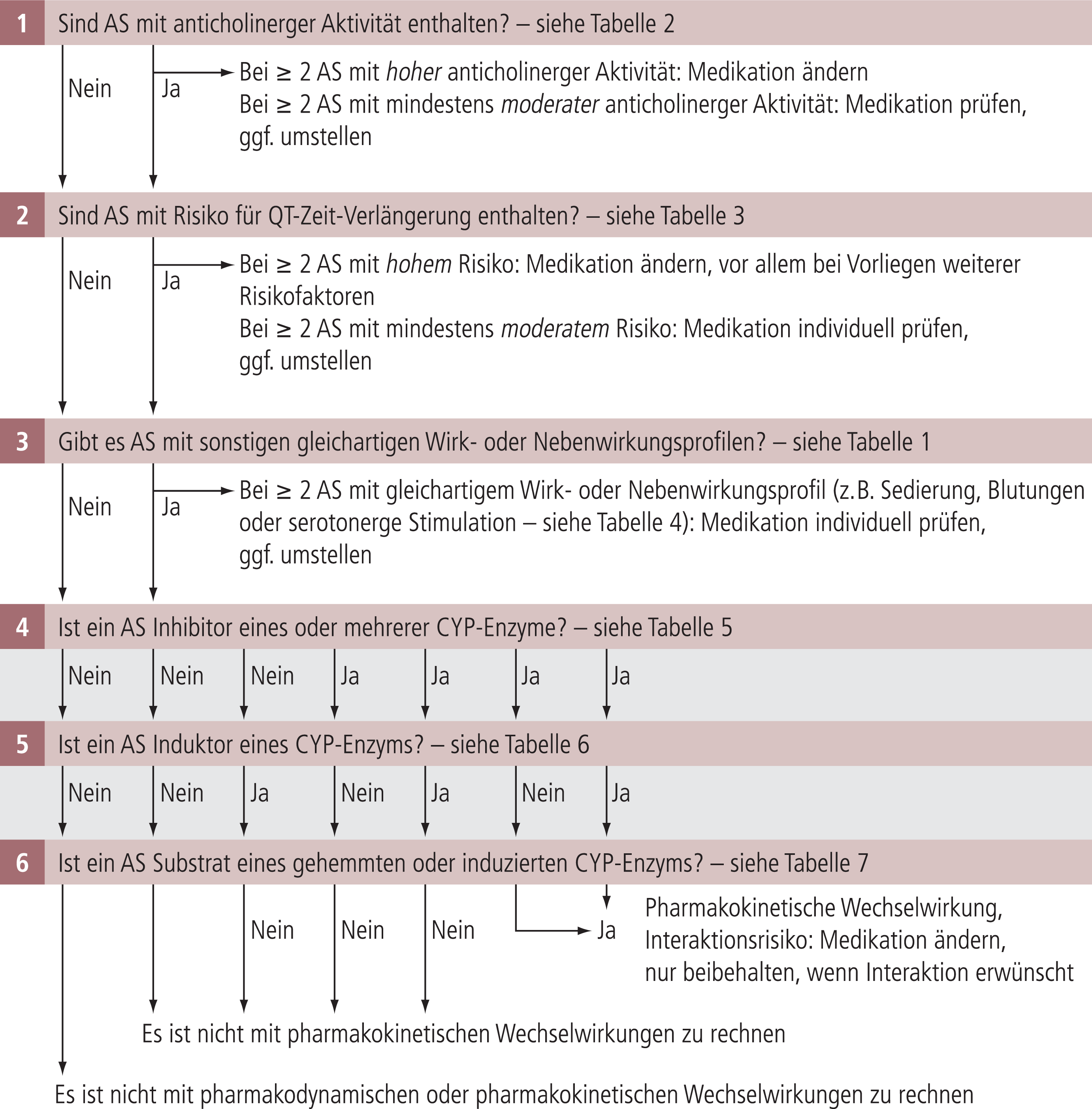
Abb. 1. Algorithmus für einen systematischen Interaktionscheck bei Kombinationsbehandlungen/Polypharmazie. Durch sequenzielle Abfrage wird überprüft, ob potenziell klinisch relevante pharmakodynamische (1.–3.) und pharmakokinetische (4.–6.) Wechselwirkungen zu erwarten sind, die zur Risikominimierung eine Änderung der Medikation erfordern. Wenn nur nach pharmakokinetischen Wechselwirkungen gesucht wird, beginnt die Abfrage bei 4. Nicht einbezogen wurden in den Interaktionscheck Wechselwirkungen mit Lithium, die die Exkretionsphase betreffen. Kritisch sind vor allem Kombinationen von Lithium mit ACE-Hemmern (Angiotensin-Konversionsenzymhemmern), nichtsteroidalen Antiphlogistika und Diuretika, die zu einem Anstieg der Wirkspiegel von Lithium und daraus folgend einer Intoxikation führen können [2]. AS: Arzneistoffe; CYP: Cytochrom-P450
Im ersten Schritt wird nach möglichen anticholinergen Interaktionseffekten gesucht. Dazu wird geprüft, ob die Liste Arzneistoffe mit hoher oder moderater anticholinerger Aktivität enthält (Tab. 2). Wenn zwei oder mehr Arzneistoffe mit hoher Aktivität identifiziert werden, dann sollte die Medikation geändert werden. Wenn zwei oder mehr Arzneistoffe moderate anticholinerge Aktivität besitzen, sollte individuell entschieden werden, ob eine Änderung vorgenommen wird.
Im zweiten Schritt wird eine mögliche Verlängerung der QT-Zeit überprüft (Tab. 3). Bei Verordnung von zwei oder mehr Arzneistoffen mit hohem Risiko sollte die Medikation geändert werden. Und es muss individuell geprüft werden, ob weitere Risikofaktoren vorliegen: Long-QT-Syndrom, Hypokaliämie, Hypomagnesiämie, akuter Myokardinfarkt, Bradykardie, Sepsis, Einnahme von Schleifendiuretika, hohe Wirkspiegel, hohe Dosis, weibliches Geschlecht [41].
Im dritten Schritt wird nach möglichen weiteren pharmakodynamischen Interaktionen gesucht, indem geprüft wird, ob Substanzen mit gleich- oder gegensinnig ausgerichtetem Wirkprofil in der Arzneimittelliste enthalten sind (Tab. 1). Dabei sollten gegebenenfalls die Fachinformationen zu Rate gezogen werden, wenn keine ausreichende Kenntnis über das Nebenwirkungspotenzial einzelner Wirkstoffe vorhanden ist. Besonders sollte auch auf mögliche serotonerge Interaktionen oder Blutbildschädigungen oder unkontrollierte Blutungen geachtet werden (Tab. 4). Solche Kombinationen sollten möglichst vermieden werden. Falls ihr Einsatz unumgänglich erscheint, ist auf engmaschige Kontrollen der klinischen Symptomatik und des Blutbilds zu achten und der Patient muss über die möglichen Symptome aufgeklärt sein.
Für die Identifizierung relevanter pharmakokinetischer Wechselwirkungen wird geprüft, ob die Liste der kombinierten Arzneimittel relevante Inhibitoren (Tab. 5) oder Induktoren (Tab. 6) enthält. Nur wenn dies zutreffend ist, muss im nächsten Schritt geprüft werden, ob sich unter den kombinierten Arzneistoffen solche befinden, die bevorzugt durch ein einzelnes CYP-Enzym metabolisiert werden (Tab. 7). Zur Risikominimierung sollten Inhibitoren (Tab. 5) oder Induktoren (Tab. 6) mit Arzneistoffen der Tabelle 7 nur dann kombiniert werden, wenn mit keiner pharmakokinetischen Wechselwirkung zu rechnen ist, oder wenn die Wechselwirkung erwünscht ist und therapeutisch genutzt werden soll.
Die in Abbildung 1 vorgeschlagene Vorgehensweise wirkt auf den ersten Blick aufwendig, vielen sicher zu aufwendig für den klinischen Alltag. Der Autor dieses Beitrags regt an, den Algorithmus bei Patienten unter Polypharmazie mit und ohne Interaktionsprobleme zu testen. Dann wird der Anwender feststellen, dass man mit den Tabellen und dem Algorithmus klinisch relevante Arzneimittelinteraktionen von Psychopharmaka aufspüren kann. Und er wird auch feststellen, dass eine Vorgehensweise beschrieben wurde, die versierte Verordner von Kombinationsbehandlungen anwenden, wenn sie möglichst sicher kombinieren wollen.
Fazit
Interaktionen sind in der Psychopharmakotherapie bei Kombinationen hoch relevant, einerseits vielfach nützlich, aber oft ein Problem der Arzneimittelsicherheit. Die Forschung der vergangen Jahre hat wesentlich dazu beigetragen, dass Arzneimittelwechselwirkungen verstehbar geworden sind. Es ist erkannt worden, dass und wie man aus dem Rezeptor- und Wirkungsprofil kombinierter Arzneistoffe pharmakodynamische Interaktionen vorhersagen kann. Aus Substrat-, Hemm- und Induktoreigenschaften der kombinierten Arzneistoffe lassen sich pharmakokinetische Wechselwirkungen vorhersagen. Daher werden heute immer weniger Interaktionen zufällig entdeckt. Verbessertes Wissen und die zunehmende Verbreitung von elektronischen Datenbanken haben dazu beigetragen, Kombinationsbehandlungen sicherer als früher zu handhaben. Wegen der enormen Kombinationsvielfalt kommt man aber nicht umhin, jede Kombinationstherapie und jede Polypharmazie individuell bezüglich Interaktionsrisiken zu überprüfen. Um die Prüfung überschaubar und machbar zu gestalten und um klinisch relevante Wechselwirkungen zu erfassen, die zu schwerwiegenden UAW führen könnten, müssen interaktionsträchtige Arzneistoffe und möglichst auch solche mit geringer therapeutischer Breite identifiziert werden. Dies kann anhand der pharmakologischen Eigenschaften der Arzneistoffe erfolgen, wie hier dargestellt. Nicht vergessen sollte man dabei, was hier nicht thematisiert wurde, dass bei Polypharmazie immer wieder kritisch hinterfragt werden muss, ob wirklich alle kombinierten Arzneimittel für die Behandlung benötigt werden.
Interessenkonflikterklärung
CH gibt als potenzielle Interessenkonflikte die folgenden Firmen und Gesellschaften an: Astra Zeneca, Esparma, Janssen-Cilag, Lilly, Lundbeck, Otsuka, Pfizer, Psiac, Servier
GE gibt als potenzielle Interessenkonflikte die folgenden Firmen und Gesellschaften an: Astra Zeneca, Janssen-Cilag, Novartis, Pfizer, Psiac, Servier, Teva
Literatur
1. Babiak W. Case fatality due to overdosage of a combination of tranylcypromine (Parnate) and imipramine (Tofranil). Can Med Assoc J 1961;85:377.
2. Benkert O, Hippius H. Kompendium der Psychiatrischen Pharmakotherapie. 9. Auflage, Heidelberg, Berlin: Springer, 2013.
3. Burrows GD, Norman TR, Marriott PF, Davies B. Zimeldine in depressive illness – efficacy and safety data. Acta Psychiatr Scand Suppl 1983;308:31–40.
4. Cascorbi I. Drug interactions – principles, examples and clinical consequences. Dtsch Arztebl Int 2012;109:546–55.
5. Chakos M, Patel JK, Rosenheck R, Glick ID, et al. Concomitant psychotropic medication use during treatment of schizophrenia patients: longitudinal results from the CATIE study. Clin Schizophr Relat Psychoses 2011;5:124–34.
6. Chermá MD, Löfgren UB, Almkvist G, Hallert C, et al. Assessment of the prescription of antidepressant drugs in elderly nursing home patients: a clinical and laboratory follow-up investigation. J Clin Psychopharmacol 2008;28:424–31.
7. Chew ML, Mulsant BH, Pollock BG, Lehman ME, et al. Anticholinergic activity of 107 medications commonly used by older adults. J Am Geriatr Soc 2008;56:1333–41.
8. Ciraulo DA, Shader RI. Fluoxetine drug-drug interactions: I. Antidepressants and antipsychotics. J Clin Psychopharmacol 1990;10:48–50.
9. de Leon J. Paying attention to pharmacokinetic and pharmacodynamic mechanisms to progress in the area of anticholinergic use in geriatric patients. Curr Drug Metab 2011;12:635–46.
10. Fagius J, Osterman PO, Sidén A, Wiholm BE. Guillain-Barré syndrome following zimeldine treatment. J Neurol Neurosurg Psychiatry 1985;48:65–9.
11. Faries D, Ascher-Svanum H, Zhu B, et al. Antipsychotic monotherapy and polypharmacy in the naturalistic treatment of schizophrenia with atypical antipsychotics. BMC Psychiatry 2005;5:26.
12. Frechen S, Zoeller A, Ruberg K, Voltz R, et al. Drug interactions in dying patients: a retrospective analysis of hospice inpatients in Germany. Drug Saf 2012;35:745–58.
13. Girardin FR, Gex-Fabry M, Berney P, Shah D, et al. Drug-induced long QT in adult psychiatric inpatients: the 5-year cross-sectional ECG screening outcome in psychiatry study. Am J Psychiatry 2013;170:1468–76.
14. Grözinger M, Dragicevic A, Hiemke C, Shams M, et al. Melperone is an inhibitor of the CYP2D6 catalyzed O-demethylation of venlafaxine. Pharmacopsychiatry 2003;36:3–6.
15. Grözinger M, Härtter S, Hiemke C, Röschke J. Oxybutynin enhances the metabolism of clomipramine and dextrorphan possibly by induction of a cytochrome P450 isoenzyme. J Clin Psychopharmacol 1999;19:287–9.
16. Guest EJ, Rowland-Yeo K, Rostami-Hodjegan A, Tucker GT, et al. Assessment of algorithms for predicting drug-drug interactions via inhibition mechanisms: comparison of dynamic and static models. Br J Clin Pharmacol 2011;71:72–87.
17. Haen E. Arzneimittelinteraktionen. Interaktionen zwischen körperfremden Substanzen. Nervenarzt 2014;85:417–26.
18. Härtter S, Dingemanse J, Baier D, Ziegler G, et al. Inhibition of dextromethorphan metabolism by moclobemide. Psychopharmacology (Berl) 1998;135:22–6.
19. Härtter S, Wetzel H, Hammes E, Hiemke C. Inhibition of antidepressant demethylation and hydroxylation by fluvoxamine in depressed patients. Psychopharmacology 1993;110:302–28.
20. Hausner H, Wittmann M, Hajak G, Haen E. Polypharmazie als geschlechtsspezifisches Phänomen in der Psychiatrie. Psychopharmakotherapie 2008;15:21–3.
21. Hiemke C, Baumann P, Bergemann N, Conca A, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry 2011;44:195–235.
22. Hiemke C, Weigmann H, Härtter S, Dahmen N, et al. Elevated levels of clozapine in serum after addition of fluvoxamine. J Clin Psychopharmacol 1994;14:279–81.
23. http://medicine.iupui.edu/clinpharm/ddis/main-table/ (letzter Zugriff am 29.09.14).
24. www.apotheken-umschau.de/Medikamente/Wechselwirkungs-Check-104131.html (letzter Zugriff am 29.09.14).
25. www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (letzter Zugriff am 29.09.14).
26. www.crediblemeds.org/ (letzter Zugriff am 29.09.14).
27. Huang J, Niu C, Green CD, Yang L, et al. Systematic prediction of pharmacodynamic drug-drug interactions through protein-protein-interaction network. PLoS Comput Biol 2013;9:e1002998.
28. Juurlink DN, Mamdani M, Kopp A, Laupacis A, et al. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA 2003;289:1652–8.
29. Kane FJ Jr, Taylor TW. A toxic reaction to combined Elavil-Librium therapy. Am J Psychiatry 1963;119:1179–80.
30. Lahdelma L, Appelberg B. Clozapine-induced agranulocytosis in Finland, 1982–2007: long-term monitoring of patients is still warranted. J Clin Psychiatry 2012;73:837–42.
31. Launiainen T, Vuori E, Ojanperä I. Prevalence of adverse drug combinations in a large post-mortem toxicology database. Int J Legal Med 2009;123:109–15.
32. Lemberger L, Rowe H, Bosomworth JC, Tenbarge JB, et al. The effect of fluoxetine on the pharmacokinetics and psychomotor responses of diazepam. Clin Pharmacol Ther 1988;43:412–9.
33. Lertxundi U, Domingo-Echaburu S, Hernandez R, Peral J, et al. Expert-based drug lists to measure anticholinergic burden: similar names, different results. Psychogeriatrics 2013;13:17–24.
34. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol 2013;27:327–36.
35. Marusic S, Bac ic-Vrca V, Obreli Neto PR, Franic M, et al. Actual drug-drug interactions in elderly patients discharged from internal medicine clinic: a prospective observational study. Eur J Clin Pharmacol 2013;69:1717–24.
36. Sharifi H, Hasanloei MA, Mahmoudi J. Polypharmacy-induced drug-drug interactions; threats to patient safety. Drug Res (Stuttg). 2014 Feb 5. [Epub ahead of print].
37. Stöllberger C, Huber JO, Finsterer J. Antipsychotic drugs and QT prolongation. Int Clin Psychopharmacol 2005;20:243–51.
38. Talbot DR. Are tranquilizer combinations more effective than a single tranquilizer? Am J Psychiatry 1964;121:597–600.
39. Tisdale JE, Jaynes HA, Kingery JR, Mourad NA, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013;6:479–87.
40. van Noord C, Eijgelsheim M, Stricker BH. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol 2010;70:16–23.
41. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J 2013;13:533–40.
42. Wenzel-Seifert K, Ostermeier CP, Ben Omar N, Haen E. Unerwünschte kardiovaskuläre Wirkungen von Psychopharmaka. Pathophysiologie und Risikominimierung. Psychopharmakotherapie 2013;20:148–57.
43. Witton K. Severe toxic reaction to combined amitriptyline and thoridazine. Am J Psychiatry 1965;121:812–3.
44. www.MediQ.ch (letzter Zugriff am 29.09.14).
45. www.psiac.de (letzter Zugriff am 29.09.14).
46. Xiang YT, Dickerson F, Kreyenbuhl J, Ungvari GS, et al. Common use of antipsychotic polypharmacy in older Asian patients with schizophrenia (2001–2009). J Clin Psychopharmacol 2012;32:809–13.
47. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013;138:103–41.
48. Zarate CA Jr, Quiroz JA. Combination treatment in bipolar disorder: a review of controlled trials. Bipolar Disord 2003;5:217–25.
49. Zhou S-F, Zhou Z-W, Li C-G, et al. Identification of drugs that interact with herbs in drug development. Drug Discovery Today 2007;2:664–73.
Prof. Dr. rer. nat. Christoph Hiemke, Klinik für Psychiatrie und Psychotherapie der Universität Mainz, Untere Zahlbacher Straße 8, 55131 Mainz, E-Mail: hiemke@uni-mainz.de
Dr. med. Gabriel Eckermann, Klinik für Psychiatrie und Psychotherapie, Bezirkskrankenhaus Kaufbeuren. Kemnater Straße 16 , 87600 Kaufbeuren, E-Mail: eckermann.iapkf@online.de
Combination therapy and polypharmacy in psychiatry and drug-drug interactions
Drug combinations and polypharmacy with five or more drugs are common and often necessary in every day pharmacotherapy to achieve best possible treatment outcomes. Treatment with several drugs, however, enhances the risk of adverse drug reactions (ADR) and drug-drug interactions. In psychiatry, many pharmacodynamic and pharmacokinetic drug interactions were discovered during the last decades by accident. Since marked advances in knowledge have resulted, drug interactions are now largely predictable and most interaction-related ADR considered as preventable medication errors. To prevent interaction-related ADR, there is extensive literature. Most helpful are computer-assisted or internet based drug interaction programs. Nevertheless, valid interaction checks are still difficult, especially to assess whether an indicated potential interaction risk is clinically relevant or not. Moreover, the combination diversity is so great under polypharmacy that it is essential to examine each administered list of prescribed drugs with respect to their interaction risk individually. For this publication, psychotropic and non-psychotropic drugs were graded according to their interaction risk burden, and an algorithm is proposed for identification of expected pharmacodynamic and pharmacokinetic interactions in a prescribed drug list of individual patients. Graduated interaction risk evaluation should be applied to decide whether a change in planned medication seems necessary or not. A change is highly recommended whenever a high risk is identified and alternative medications without interaction risk are available.
Key words: Drug-drug interactions, pharmacokinetic, pharmacodynamic, anticholinergic, QT-time, adverse drug reactions
Psychopharmakotherapie 2014; 21(06)