Holger Petri, Bad Wildungen
„Analysen von Wechselwirkungen: Großer Aufwand – k(l)eine Wirkung?“, so betitelt Der Arzneimittelbrief einen Beitrag zur Überprüfung von Arzneimittelinteraktionen [1]. Der Artikel beschreibt, dass die Suche nach klinisch relevanten Wechselwirkungen mit zunehmender Zahl der verordneten Arzneimittel derart umfangreich wird, dass eine Interaktionsanalyse ohne elektronische Systeme kaum bewältigt werden kann. Da es bei der Analyse der Wechselwirkungen keine standardisierte Vorgehensweise gibt, ist die Leistungsfähigkeit der angebotenen Systeme sehr unterschiedlich und die Ergebnisse sind somit weitgehend inkonsistent. Problematisch bei der Erfassung von Arzneimittelinteraktionen mit einer Software ist außerdem, dass häufig irrelevante Warnmeldungen abgegeben werden: Von 100 potenziellen Interaktionen werden letztlich nur 5 bis 10 klinisch relevant. Dieser „Alert Overkill“ stört den praktischen Arbeitsablauf in der medizinischen Versorgung der Patienten („Zeitklau“ und „Therapieverschleppung“). Dazu kommt, dass die Programme keine alternativen Arzneimittel vorschlagen [1].
Lösungsansatz
Enzyme der Cytochrom-P450-(CYP450-)Familie sind häufig an Phase-I-Reaktionen beteiligt und besitzen größte Bedeutung in der Biotransformation von Arzneistoffen. Zudem können Arzneimittel die Enzymaktivität hemmen oder induzieren [7]. Für das Verständnis von Wechselwirkungen ist es daher wichtig, Kenntnisse über die gegenseitige Beeinflussung von Arzneistoffen und CYP450-Isoenzymen zu erwerben.
Anhand der Interaktionstabellen in dieser Serie wird dargestellt, ob pharmakologisch vergleichbare Wirkstoffe in klinisch relevantem Ausmaß Substrate bestimmter CYP450-Isoenzyme sind und ob sie modulierende Eigenschaften gegenüber diesen Enzymen besitzen. Die Tabelle gibt Hinweise zum Interaktionsrisiko und damit verbunden unterschiedlich gewichtete Empfehlungen zur Nutzung einer Interaktionsdatenbank. Die Empfehlungen berücksichtigen auch pharmakodynamische Parameter wie den therapeutischen Index [11], eine potenzielle QTc-Zeit-Verlängerung oder eine hohe Affinität zu Acetylcholin- und Serotonin-Rezeptoren des Wirkstoffs, die ihn „anfällig“ für klinisch relevante Wechselwirkungen machen. Weiterhin enthält die Interaktionstabelle wirkstoffspezifische Informationen und Angaben zur Häufigkeit von pharmakogenetischen Polymorphismen einzelner Cytochrome. In diesem ersten Beitrag der Serie wird das Interaktionspotenzial niederpotenter Neuroleptika vorgestellt (Tab. 1).
In Abbildung 1 sind Substanzen mit mittelstarker bis starker inhibitorischer bzw. induzierender Wirkung auf klinisch bedeutsame CYP450-Isoenzyme zusammengetragen. Eine farbliche Unterlegung in Abbildung 1 und Tabelle 1 erleichtert die Zuordnung von Enzym und Substanzen und zeigt somit diejenigen Kombinationspaare, die zu einer klinisch relevanten Wechselwirkung führen können und daher einer speziellen Überwachung, einer mithilfe des therapeutischen Drug-Monitorings (TDM) gesteuerten Dosisanpassung oder therapeutischer Alternativen bedürfen. Alternativ zum Einsatz kommende Wirkstoffe können der Interaktionstabelle entnommen werden.
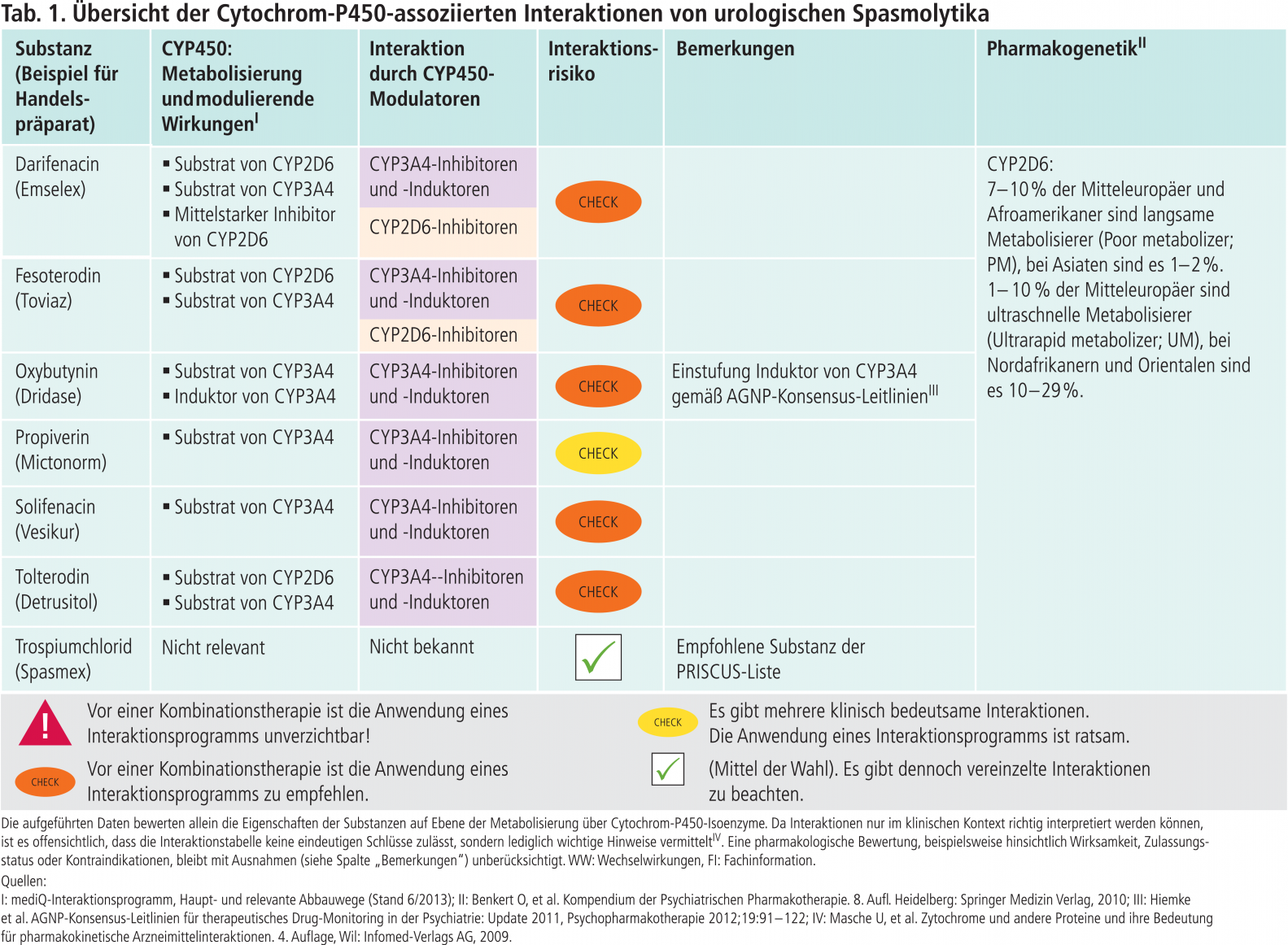
Tab. 1. Übersicht der Cytochrom-P450-assoziierten Interaktionen niederpotenter Neuroleptika (Stand: 11/2012).
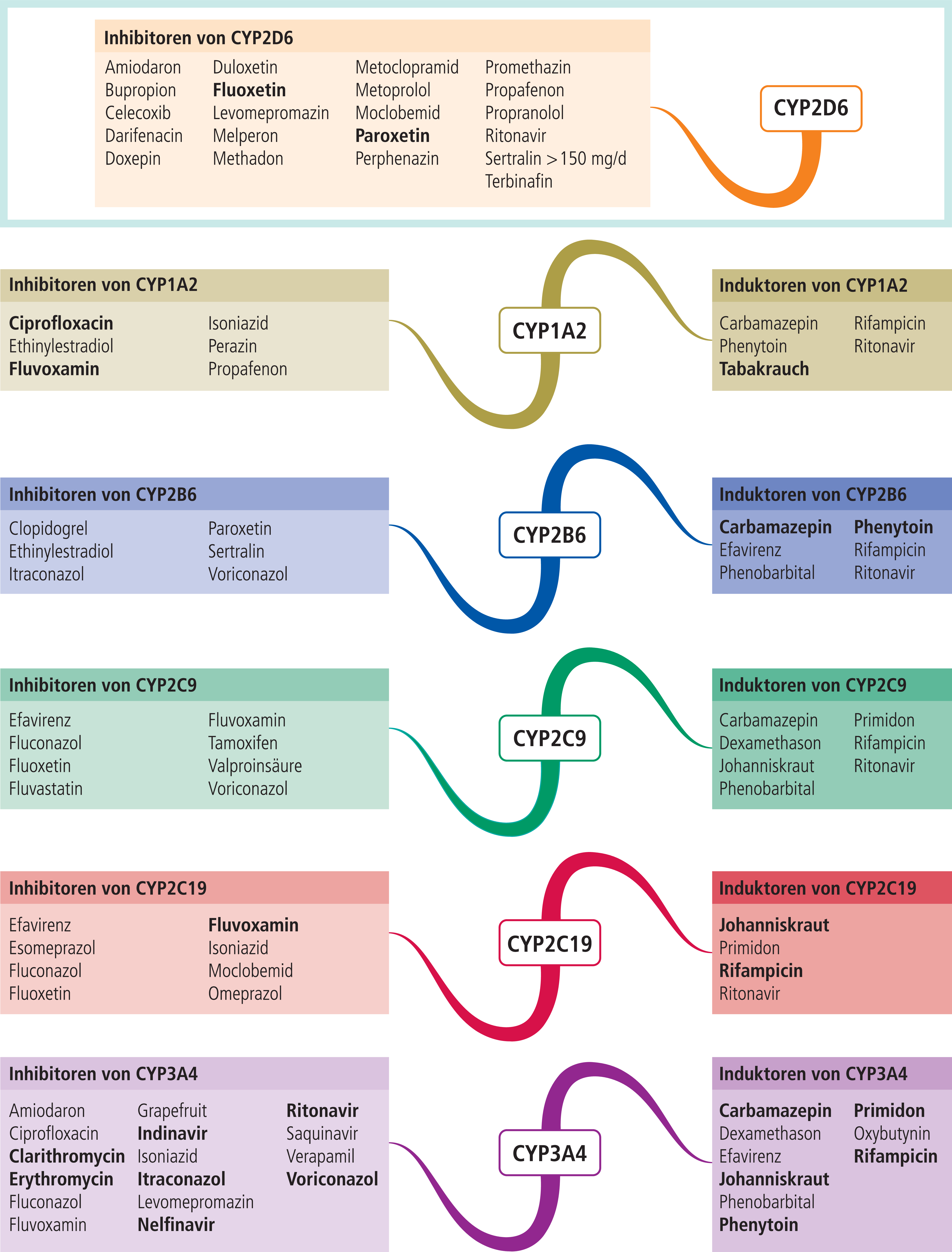
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf einzelne CYP450-Isoenzyme (Stand: 11/2012) [Quelle: mediQ-Interaktionsprogramm].
Ist das Interaktionsrisiko auf Ebene der Cytochrome insgesamt niedrig, so hat dies den zusätzlichen Vorteil, dass Medikamente später wieder abgesetzt werden können, ohne bei der bestehenden Medikation signifikante Änderungen der Wirkstoffspiegel und damit eine Gefährdung der Therapie bzw. des Patienten zu riskieren. Beispielsweise senkt Carbamazepin über eine Induktion des CYP450-Isoenzyms 3A4 die AUC (Fläche unter der Konzentrations-Zeit-Kurve) von Simvastatin um etwa 75%. Dies kann nach Monitoring der Blutfettwerte eine Dosiserhöhung von Simvastatin notwendig machen [13]. Wird Carbamazepin abgesetzt, steigen nach der Deinduktionszeit die Wirkstoffspiegel von Simvastatin. Falls die Dosis des CSE-Hemmers nicht reduziert wird, ist folglich das Risiko für eine Myopathie bzw. Rhabdomyolse erhöht. Wird statt Simvastatin alternativ Pravastatin verordnet, ist keine klinisch relevante Interaktion mit Carbamazepin zu erwarten.
Bleibt eine medikamentöse Therapie dennoch erfolglos oder kommt es zu toxischen Effekten und eine Wechselwirkung mit anderen Arzneimitteln ist auszuschließen, könnte das auf eine unregelmäßige Einnahme der Medikamente oder auf eine Stoffwechselstörung aufgrund einer genetischen Variation hindeuten.
Methode
Das Wissen um pharmakokinetische Wechselwirkungen auf Ebene der CYP450-Isoenzyme ist in den letzten Jahren enorm gewachsen [4]. Die Qualität und somit die Aussagekraft der Studien ist aufgrund fehlender einheitlicher Standards unterschiedlich.
Die Autoren der für die Angaben in diesem Beitrag dienenden Referenzdatenbank, mediQ, Schweiz, teilen die modulierenden Eigenschaften der Substanzen in drei Klassen ein:
- Bei In-vivo-Daten in Abhängigkeit von klinisch relevanten Veränderungen meist in die Stufen 2 und 3,
- bei In-vitro-Daten und keinen oder zweifelhaften Hinweisen auf eine In-vivo-Wirkung in Stufe 1, es sei denn, es handelt sich um eine neue Substanz und die Autoren der Studien spekulieren auf eine klinisch relevante Wechselwirkung [10].
Die Abbauwege werden laut mediQ wie folgt eingestuft:
- Ein Hauptabbauweg liegt vor, wenn es sich um den einzigen Abbauweg handelt oder wenn im Fall der Beeinträchtigung dieses Abbauweges eine Wirkung mit hoher klinischer Relevanz resultiert.
- Ein relevanter Abbauweg liegt vor, wenn durch dessen Beeinträchtigung eine klinisch relevante Veränderung erfolgt, die aber in der Regel keine hohe klinische Bedeutung hat.
- Nebenwege sind ohne klinische Relevanz, es sei denn, ein anderer relevanter Abbauweg fällt aus, beispielsweise bei langsamen Metabolisierern (Poor metabolizer) eines bestimmten Enzyms.
Für die Angaben in Tabelle 1 wurden aus mediQ die Hauptabbauwege und die relevanten Abbauwege identifiziert.
Zum Abgleich der Angaben zu den modulierenden Eigenschaften wurde als zweite Datenbank GeneMedRx, USA, genutzt. Die Autoren dieser Datenbank orientieren sich bei der Einstufung an der FDA-Klassifikation der prozentualen Veränderung von AUC und Clearance, unabhängig vom Studientyp [9]. Bei einem Unterschied von zwei Stufen wurden die Autoren von mediQ um Stellungnahme gebeten und die rückgemeldeten Daten für die Erstellung von Abbildung 1 berücksichtigt. In Korrespondenz mit Prof. Christoph Hiemke, Mainz, TDM-Gruppe der Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP), sind aus dieser Abbildung entbehrliche Angaben gestrichen worden, da sie keine klinische Relevanz haben und so zu Overalerting führen würden. Hiervon betroffen waren die CYP-Inhibitoren Clobazam, Haloperidol, Hydroxyzin (2D6), Tamoxifen (3A4) und Estradiol (1A2). Zu Sertralin wurde der Vermerk „>150 mg/d“ ergänzt. Das Blasenspasmolytikum Oxybutynin wurde als CYP3A4-Induktor der Tabelle hinzugefügt.
Auf obsolete Substanzen wurde zugunsten der Übersichtlichkeit der Tabellen verzichtet. Auch onkologische Arzneimittel blieben unberücksichtigt, da ein Interaktionscheck bei diesen selbstverständlich sein sollte. Die finale Version (Abb. 1) bietet damit eine übersichtliche Zusammenfassung von Modulatoren.
Diskussion
Mithilfe von Tabellen und Abbildungen mit einfach verständlicher Symbolik soll den eingangs geschilderten Nachteilen von elektronischen Systemen zur Analyse des Interaktionspotenzials gleichzeitig verabreichter Medikamente auf pragmatische Art begegnet werden. Arzt und Apotheker können sich damit einen schnellen Überblick darüber verschaffen, bei welchen Arzneimittelkombinationen mit klinisch relevanten Wechselwirkungen zu rechnen ist. Durch die Gegenüberstellung pharmakologisch vergleichbarer Wirkstoffe bietet sich dem Arzt die Möglichkeit, bei der Verordnung diejenigen Substanzen auszuwählen, die wenig anfällig für CYP450-bedingte Wechselwirkungen sind.
Wird gemäß Tabelle 1 ein Interaktionsprogramm zur Anwendung empfohlen, so sollte dieses neben den Fachinformationen bzw. SPCs (Summaries of product characterics) auch andere Quellen wie Studien und die wesentlichen Datenbanken zur Einschätzung des Wechselwirkungsrisikos heranziehen und dem Stand der Wissenschaft entsprechen. Wünschenswert ist, dass die Empfehlung des Interaktionsprogramms die berücksichtigten Originalarbeiten als Quellen angibt.
In der Regel erfolgen pharmakokinetische Studien mit Substanzpaaren bestehend aus einem Substrat und einer modulierenden Substanz. Erhält der Patient fünf oder mehr unterschiedliche Wirkstoffe (Polypharmazie), ist die Wahrscheinlichkeit groß, dass mehr als nur eine Substanz inhibierend bzw. induzierend auf das jeweilige CYP450-Isoenzym wirkt. Wie sich dies wiederum auf den Wirkstoffspiegel des betreffenden Substrates auswirkt, kann ohne TDM nur geschätzt werden. Im Einzelfall sollte der Patient daher speziell überwacht oder auf alternative Medikamente umgestellt werden.
Mit Stand August 2012 sind in der Datenbank mediQ über 26000 Kombinationen beschrieben; rechnerisch kommt man bei einer Zahl von 2000 Substanzen auf 2 Millionen Kombinationen (Anzahl möglicher Wechselwirkungen: n · [n–1]/2; n: Anzahl Substanzen). Wünschenswert ist ein Interaktionsprogramm mit Online-Beratung (z.B. mediQ, Psiac), um aktuell vorliegende Kombinationen prüfen zu lassen.
Viele Patienten haben unter klinisch üblichen Dosierungen ungewöhnlich hohe oder niedrige Wirkstoffspiegel, weil bei ihnen die Metabolisierungsgeschwindigkeit verändert ist. Dies kann durch Komedikation verursacht oder angeboren sein [2]. So berichtete kürzlich die US-amerikanische Zulassungsbehörde FDA über drei Todesfälle nach Einnahme von Codein bei Kindern im Alter von 2 bis 5 Jahren. In allen drei Fällen wurde Codein unter Beachtung der empfohlenen Dosierung zur Schmerzbehandlung eingesetzt. Codein ist ein Prodrug, das erst durch Bioaktivierung über das Enzym CYP2D6 zur Wirkform Morphin metabolisiert wird. Aufgrund eines genetischen Polymorphismus waren alle drei verstorbenen Kinder ultraschnelle Metabolisierer, so dass das Codein bei ihnen sehr schnell zu Morphin umgewandelt wurde, wodurch es zu einer ausgeprägten Atemdepression mit Todesfolge kam [8].
Tabelle 1 informiert daher zusätzlich über die Häufigkeit möglicher Genvarianten einzelner CYP-Isoenzyme.
Die Enzymaktivität und damit die Metabolisierungsgeschwindigkeit kann aber auch mit zunehmendem Alter sinken, durch Nieren- und Lebererkrankungen [5] und in der Schwangerschaft [6] verändert sein. Ebenso unterdrücken Zytokine wie der Entzündungsmediator Interleukin-6 die Bildung hepatischer Cytochrome [3].
Die pharmakokinetische Variabilität ist somit ein häufiger Grund für unzureichendes Therapieansprechen oder für eine schlechte Verträglichkeit von Medikamenten [4]. Außerdem haben bestimmte Lebensgewohnheiten Einfluss auf die Metabolisierung. Bei Rauchern liegen die Serumspiegel von Olanzapin beispielsweise um 50% niedriger als bei Nichtrauchern. Tabakrauch induziert das Enzym CYP1A2, das für den Abbau des atypischen Neuroleptikums hauptverantwortlich ist [12].
Das Wissen über das Verhalten von Arzneistoffen gegenüber CYP450-Isoenzymen reduziert das Risiko für ein Therapieversagen bzw. für toxische Wirkungen. Im Einzelfall kann erwogen werden, Medikamente aus dem Ausland zu importieren, wenn ein Patient eine besonders interaktionsträchtige Medikation verordnet bekommt und in Deutschland hinsichtlich der CYP-bedingten Wechselwirkungen keine risikoarme Alternative zur Verfügung steht. Soll beispielsweise bei einem Patienten, der eine Therapie mit dem Proteasehemmer Ritonavir erhält, zusätzlich ein Antidepressivum verschrieben werden, ist das SNRI Milnacipran gegenüber den in Deutschland zugelassenen Standardwirkstoffen von Vorteil, da es unabhängig von CYP-Isoenzymen verstoffwechselt wird und daher keine pharmakokinetischen Wechselwirkungen zu befürchten sind.
Erwartungsgemäß erhöht sich mit der Zahl an eingesetzten Wirkstoffen die Gefahr für pharmakokinetische und pharmakodynamische Wechselwirkungen und damit für unerwünschte Wirkungen [14]. Die in dieser Reihe vorgestellten Interaktionstabellen sollen dem Arzt und Apotheker helfen, die Medikation so zu optimieren, dass ein möglichst geringes Risiko für klinisch relevante Interaktionen, zumindest auf Ebene der Cytochrome, besteht.
Literatur
1. Analysen von Wechselwirkungen: Großer Aufwand – k(l)eine Wirkung? Arzneimittelbrief 2011;45; Ausgabe Deutschland.
2. Böhm R, Reinecke K, Haen E, Cascorbi I, Herdegen T. Arzneimittelinteraktionen. Dtsch Apo Ztg 2012;36:64–74.
3. Fachinformation Roactemra®, Stand Juni 2012.
4. Hiemke C. Therapeutisches Drug-Monitoring (TDM) in der Psychiatrie. Psychopharmakotherapie 2012;19:89–90.
5. Hiemke C, Baumann P, Bergemann N, Conca A, et al. AGNP-Konsensus-Leitlinien für therapeutisches Drug-Monitoring in der Psychiatrie: Update 2011. Psychopharmakotherapie 2012;19:91–122.
6. Masche U. Pharmakokinetische Veränderungen in der Schwangerschaft. pharma-kritik 2010;32:61–5.
7. Masche U, Gysling E, Ritzmann, P. Zytochrome und andere Proteine und ihre Bedeutung für pharmakokinetische Arzneimittelinteraktionen. 4. Auflage, Wil: Infomed-Verlags AG, 2009.
8. Mitteilung des BfArM vom 30.08.2012.
9. Persönliche Stellungnahme von D. Rustan vom 15.08.2012.
10. Persönliche Stellungnahme von E. Jaquenoud Sirot vom 26.07.2012.
11. Polasek T, Lin FP, Miners JO, Doogue MP. Perpetrators of pharmacokinetic drug-drug interactions arising from altered cytochrome P450 activity. Br J Clin Pharmacol 2011;71:727–36.
12. Rao ML, et al. Olanzapin: Pharmakologie, Pharmakokinetik und therapeutisches Drug Monitoring. Fortschr Neurol Psychiat 2001;69:510–7.
13. Suchresultat Kombination Carbamazepin und Simvastatin, mediQ, vom 18.09.2012.
14. Uhrhan T, Schaefer M. Arzneimittelversorgung und Arzneimittelsicherheit in stationären Pflegeeinrichtungen. Bundesgesundheitsblatt 2010, doi 10.1007/s00103-010-1053-8.
Nachdruck aus Krankenhauspharmazie 2013;34:15–9.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2013; 20(01)