Walter E. Müller, Frankfurt/Main, und Heinz Reichmann, Dresden
Entwicklung von Selegilin und Rasagilin
Die Entwicklung von Hemmstoffen des Enzyms Monoaminoxidase (MAO) als zentral wirksame Arzneistoffe geht auf den in den 50er-Jahren gewonnenen Zufallsbefund zurück, dass ein ursprünglich als Tuberkulostatikum entwickelter Arzneistoff (Isoniazid) bei mäßiger chemotherapeutischer Wirkung antidepressive Effekte an Patienten zeigte. Dieser Befund wurde weiter verfolgt und führte zur Einführung verschiedener Monoaminoxidase-Hemmer als Antidepressiva, von denen in Deutschland noch der unselektive und irreversible MAO-Hemmer Tranylcypromin und der reversible MAO-A-Hemmer Moclobemid übrig geblieben sind, die in der Therapie depressiver Erkrankungen eine allerdings begrenzte Bedeutung haben. Die MAO-A-Hemmung wurde sehr bald mit erheblichen peripheren unerwünschten Arzneimittelwirkungen wie Bluthochdruck, Kopfschmerzen und Herzrhythmusstörungen assoziiert, besonders wenn Monoamine wie Tyramin mit der Nahrung aufgenommen wurden. Eine MAO-A-Hemmung, vor allen Dingen im Zusammenhang mit anderen Serotonin-verstärkenden Substanzen, hat darüber hinaus eine Bedeutung für die gravierende unerwünschte Arzneimittelwirkung eines Serotonin-Syndroms. Aus diesem Grunde wurde Ende der 60er Jahre die Entwicklung von Selegilin mit großem Interesse aufgenommen, als man nämlich zeigen konnte, dass diese Substanz sehr viel stärker die Monoaminoxidase-B-Isoform als die A-Isoform in der Peripherie und im zentralen Nervensystem (ZNS) hemmt. Da im ZNS Monoaminoxidase B besonders für den oxidativen Abbau des Neurotransmitters Dopamin verantwortlich ist, lag es auf der Hand, diesen selektiven MAO-B-Inhibitor im Hinblick auf eine mögliche Wirksamkeit bei Morbus Parkinson zu untersuchen. Dies führte letztlich zur klinischen Einführung von Selegilin in die Parkinson-Therapie (z.B. Movergan®).
Selegilin hatte von Anfang an ein intrinsisches Problem, da es chemisch von den Amphetaminen abgeleitet ist und am Menschen sowie am Versuchstier in erheblichem Maße im Rahmen seines komplexen Metabolismus zu Methamphetamin und Amphetamin abgebaut wird (Abb. 1). Dieser Nachteil des Selegilins wurde in der Entwicklung des ebenfalls selektiven und irrreversiblen MAO-B-Hemmers Rasagilin umgangen, der eine Aminoindan-Grundstruktur besitzt, damit chemisch keine Analogien zu den Amphetaminen zeigt und auch am Menschen und Tier nicht zu Amphetamin-Derivaten, sondern zu 1-Aminoindan verstoffwechselt wird (Abb. 1), das als Hemmstoff der Monoaminoxidase B keine Rolle spielt. Während für den Metabolismus von Selegilin aus der Gruppe der Cytochrom-P450-Enzyme im Wesentlichen die Isoenzyme CYP2B6 und CYP2C19 von Bedeutung sind, wird der Metabolismus von Rasagilin im Wesentlichen über CYP1A2 zu Aminoindan vermittelt [73, 74] (Abb. 1).
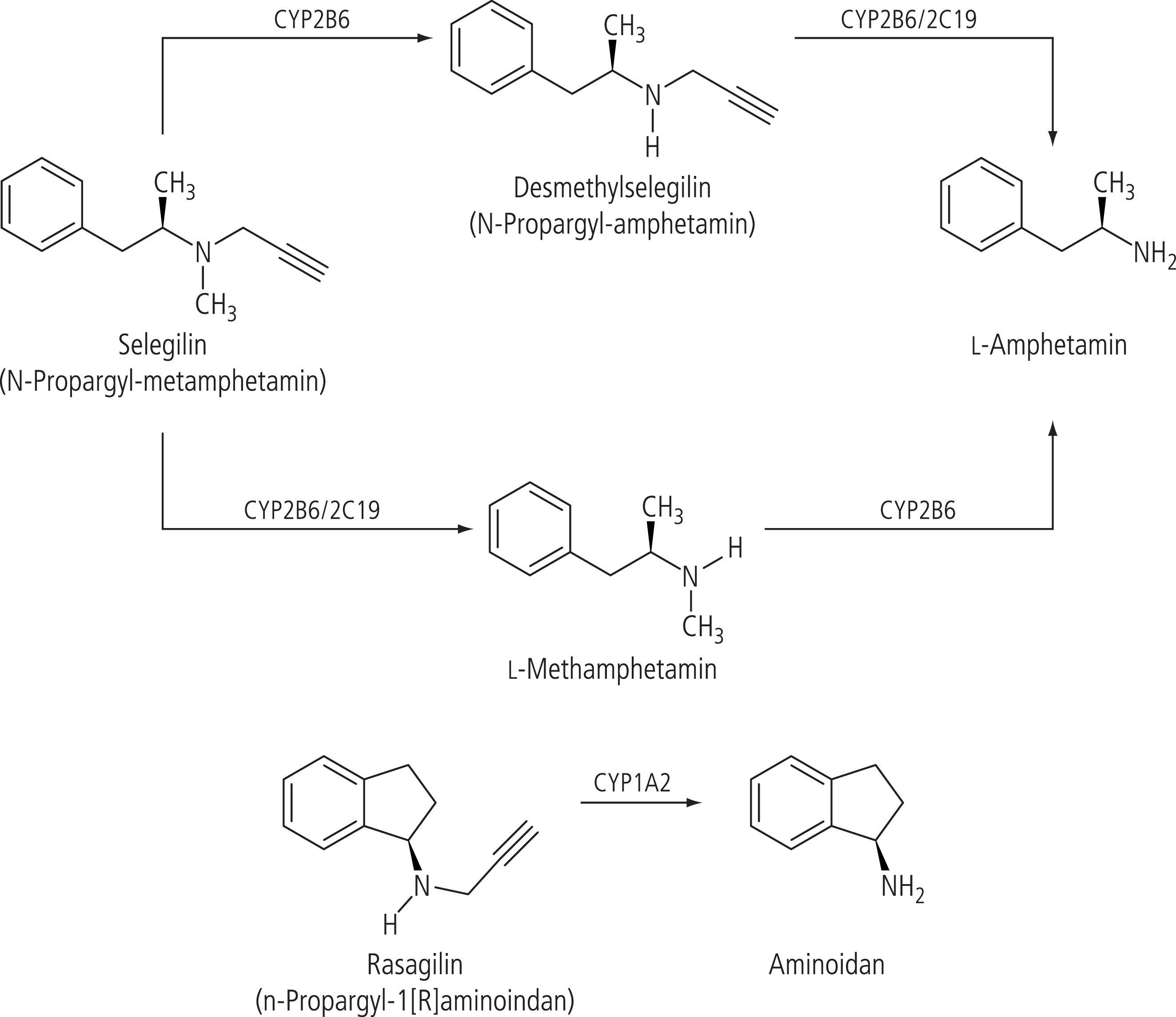
Abb. 1. Die wichtigsten Biotransformationswege von Selegilin und Rasagilin im Menschen [mod. nach 13]
Pharmakokinetik
Plasmaspiegelverläufe
Die wichtigsten Daten zur Humanpharmakokinetik beider Substanzen sind in Tabelle 1 zusammengefasst (siehe auch [44, 46]). Selegilin wird relativ schnell resorbiert und erreicht nach einer knappen Stunde maximale Plasmaspiegel. Die terminale Halbwertszeit liegt bei etwa 1,5 Stunden. Selegilin weist eine relativ hohe Gesamt-Clearance auf (knapp 60 l/min; etwa das 30fache des hepatischen Durchflusses), was als Hinweis auf einen extensiven, auch extrahepatischen Metabolismus gewertet wird [46]. Es hat ein relativ hohes Verteilungsvolumen und wird fast vollständig metabolisiert. Der unverändert im Urin ausgeschiedene Anteil ist vernachlässigbar. Die absolute Bioverfügbarkeit ist mit weniger als 10% sehr gering. Unter subchronischer Einnahme von Selegilin kommt es zu Veränderungen der Pharmakokinetik mit einer Verlängerung der Eliminationshalbwertszeit und einer ungefähren Verdoppelung der maximalen Plasmaspiegel (Tab. 2). Der pharmakologisch inaktive Metabolit Desmethylselegilin zeigt eine Eliminationshalbwertszeit von etwa 2 Stunden, während die pharmakologisch relevanten Hauptmetabolitem Methamphetamin und Amphetamin Eliminationshalbwertszeiten von rund 15 Stunden zeigen. Beide kumulieren auch deutlich bei subchronischer Therapie im Plasma, wie an der erhöhten Fläche unter der Plasmakonzentrations-Zeit-Kurve (AUC) zu erkennen ist (Tab. 2).
Tab. 1. Pharmakokinetische Eckdaten von Selegilin und Rasagilin am Menschen (gesunde Probanden)
|
Selegilin |
Rasagilin |
|
|
tmax [h] |
0,6 |
0,5 |
|
t½ [h] |
1,5 |
1,0 |
|
V [l] |
1850 |
243 |
|
Cl [l/min] |
59 |
1,6 |
|
± [%] |
9,4 |
36 |
Die Daten sind den Fachinformationen oder den Publikationen von Mahmood et al. [45]; Chen et al. [13]) entnommen. Bei Parkinson-Patienten war die mittlere t½ 1,3 Stunden [12].
tmax : Zeit maximaler Plasmaspiegel; t½: Eliminationshalbwertszeit; V: Verteilungsvolumen; Cl: Gesamte Körper-Clearance; ± : Bioverfügbarkeit
Tab. 2. Plasmaspiegel und Pharmakokinetik von Selegilin und dessen Hauptmetaboliten nach multipler Dosierung (10 mg/Tag) über 8 Tage am Menschen [nach 39]
|
Tag 1 |
Tag 8 |
|
|
Selegilin |
||
|
AUCτ [ng/ml x h]) |
1,0±0,9 |
2,7±1,8 |
|
Cmax [ng/ml] |
0,8±2,8 |
1,6±1,3 |
|
tmax [h] |
0,75 (0,5–1,5) |
0,5 (0,25–1,0) |
|
t1/2 [h] |
1,5±0,6 |
3,5±2,7 |
|
Desmethylselegilin |
||
|
AUCτ [ng/ml x h] |
31,3±7,5 |
45,7±13,8 |
|
Cmax [ng/ml] |
17,2±6,7 |
17,9±5,8 |
|
tmax [h] |
1,0 (0,5–1,5) |
0,75 (0,5–1,5) |
|
t1/2 [h] |
3,4±3,0 |
5,3±3,0 |
|
L-Methamphetamin |
||
|
AUCτ [ng/ml x h] |
161±38 |
278±92 |
|
Cmax [ng/ml] |
13,8±3,3 |
20,5±5,3 |
|
tmax [h] |
1,5 (1,0–4,0) |
1,5 (1,0–3,0) |
|
t1/2 [h] |
11,3±2,7 |
12,3±3,6 |
|
L-Amphetamin |
||
|
AUCτ [ng/ml x h] |
49,5±6,8 |
95,8±19,9 |
|
Cmax [ng/ml] |
3,3±0,4 |
6,2±1,6 |
|
tmax [h] |
2,0 (1,0–8,0) |
1,0 (1,0–8,0) |
|
t1/2 [h] |
15,8±4,6 |
13,1±4,2 |
Die Daten sind Mittelwerte±SD bzw. Bereiche von insgesamt 12 Probanden.
AUCτ: Fläche unter der Plasmakonzentrations-Zeit-Kurve im Dosierungsintervall (τ)
Die Pharmakokinetik von Selegilin wird weder durch die Nahrung noch durch das Alter der Patienten noch durch das Geschlecht wesentlich verändert [46].
Rasagilin wird ähnlich dem Selegilin nach oraler Applikation sehr schnell resorbiert und erreicht maximale Plasmawerte eine knappe Stunde nach Einnahme. Auch Rasagilin wird sehr schnell eliminiert mit einer terminalen Halbwertszeit von 1 bis 3 Stunden (Tab. 1; [12, 13, 66]).
Die Clearance von Rasagilin ist geringer als bei Selegilin und wahrscheinlich ausschließlich hepatisch. Ein Unterschied zu Selegilin ist die klare Linearität der Pharmakokinetik. Ein weiterer wesentlicher Unterschied ist die deutlich bessere Bioverfügbarkeit mit einem Wert von knapp 40%. Damit kommt es zu einem wesentlich sichereren Verhältnis von Dosis und Plasmaspiegel. Im Vergleich zu dem sehr komplexen Metabolisierungsschema des Selegilins am Menschen ist die rein hepatische Elimination des Rasagilins sehr einfach, denn die Substanz wird im Wesentlichen nur zu einem Metaboliten (1-R-Aminoindan) metabolisiert unter hauptsächlicher Beteiligung von CYP1A2 (Abb. 1). Der Metabolit zeigt weder vasoaktive- noch MAO-hemmende Effekte, spielt aber eine Rolle für die neuroprotektiven Eigenschaften (siehe unten).
Zwischen Selegilin und Rasagilin bestehen also Unterschiede im Metabolisierungsverhalten mit den pharmakologisch aktiven Selegilin-Hauptmetaboliten Methamphetamin und Amphetamin, der linearen Pharmakokinetik von Rasagilin im therapeutischen Bereich wie auch dessen deutlich besserer Bioverfügbarkeit. Bei der Bewertung der pharmakokinetischen Eckdaten von Selegilin am Menschen muss man allerdings beachten, dass durch die irreversible, sehr schnelle Hemmung des Targets Monoaminoxidase B („hit and run“-Effekt) die primäre Pharmakokinetik wie auch die Eliminationshalbwertszeit für die Therapie von untergeordneter Bedeutung sind, wie im folgenden Abschnitt dargestellt.
Zeitverlauf der MAO-B-Hemmung
Humanthrombozyten, aber auch die Thrombozyten vieler Versuchstiere exprimieren substanzielle Mengen von Monoaminoxidase B. Experimentelle Untersuchungen an Labortieren zeigten eine gute zeitliche Übereinstimmung zwischen der irreversiblen Hemmung der Monoaminoxidase B im Gehirn und auf den Plättchen durch Selegilin und Rasagilin, wobei der Effekt im Gehirn meist länger andauerte. Eine gute Korrelation zwischen MAO-B-Hemmung im Gehirn und an Thrombozyten wurde auch am Menschen gezeigt [6]. Auf der Basis dieser Befunde hat man auch für Selegilin und Rasagilin in pharmakokinetischen Untersuchungen Messungen der MAO-B-Aktivität an Humanthrombozyten benutzt, um Hinweise auf den Zeitverlauf der MAO-B-Hemmung am Patienten zu erhalten. Erste Befunde von Lee et al. [41] zeigten, dass schon nach einer Einmaldosis von 10 mg Selegilin bereits nach dem ersten Tag die MAO-B-Aktivität in Plättchen praktisch zu 100% gehemmt ist und dass dieser Effekt über die restlichen Tage der Therapie stabil bleibt. Nach Absetzen der Selegilin-Therapie kehrte die MAO-B-Aktivität der Plättchen mit einer Verzögerung von etwa 14 Tagen zu 100% Aktivität zurück, was angesichts der irreversiblen Blockade der Thrombozyten-MAO-B über die Thrombozyten-Neusynthese erklärt wurde. Diese Befunde entsprechen Daten von Simpson et al. [63]. Die Befunde für Rasagilin sind sehr ähnlich. Auch hier wurde bei subchronischer Therapie mit Rasagilin in den Dosen 2 mg, 5 mg bzw. 10 mg/Tag nach wenigen Tagen der Therapie eine praktisch 100%ige Hemmung der MAO-B in den Plättchen erreicht [66]. Auch bei Rasagilin wurde wieder eine volle MAO-B-Aktivität 14 Tage nach Absetzen der Therapie gesehen, was sehr gut mit den Daten für Selegilin übereinstimmt und sich über die Neusynthese von Thrombozyten erklären lässt [66].
Zwei PET(Positronen-Emissions-Tomographie)-Studien haben in jüngster Zeit Hinweise auf den Zeitverlauf der MAO-B-Hemmung im menschlichen Gehirn nach Einnahme von Selegilin oder Rasagilin geliefert. In der Untersuchung von Fowler et al. [27] wurde gezeigt, dass bei täglicher Einnahme von 10 mg Selegilin nach einigen Tagen eine praktisch maximale MAO-B-Hemmung (>95%) im menschlichen Gehirn erreicht werden konnte. Nach Absetzen der Selegilin-Therapie wurden rund 40 Tage benötigt, bevor die MAO-B-Aktivität zu den Ausgangswerten zurückkehrte, was mit der relativ langsamen Neusynthese von MAO-B im Gehirn [73] übereinstimmt. In einer ähnlichen Untersuchung von Freedman et al. [28] wurden ähnliche Befunde für eine tägliche Dosis von 1 mg Rasagilin gesehen. Auch hier benötigte das System rund 40 Tage, bevor durch Neusynthese der MAO-B die Ausgangswerte für die Enzymaktivität erreicht waren.
Pharmakodynamik in Bezug zum Monoaminstoffwechsel
MAO-B-Hemmung
Selegilin und Rasagilin hemmen konzentrationsabhängig die beiden Monoaminoxidase-Isoenzyme A und B. Allerdings unterscheiden sie sich im Hinblick auf ihre Potenz an beiden Isoenzymen um etwa zwei Zehnerpotenzen, was sich auch in den entsprechenden halbmaximalen Hemmkonzentrationen (IC50) niederschlägt (Abb. 2).
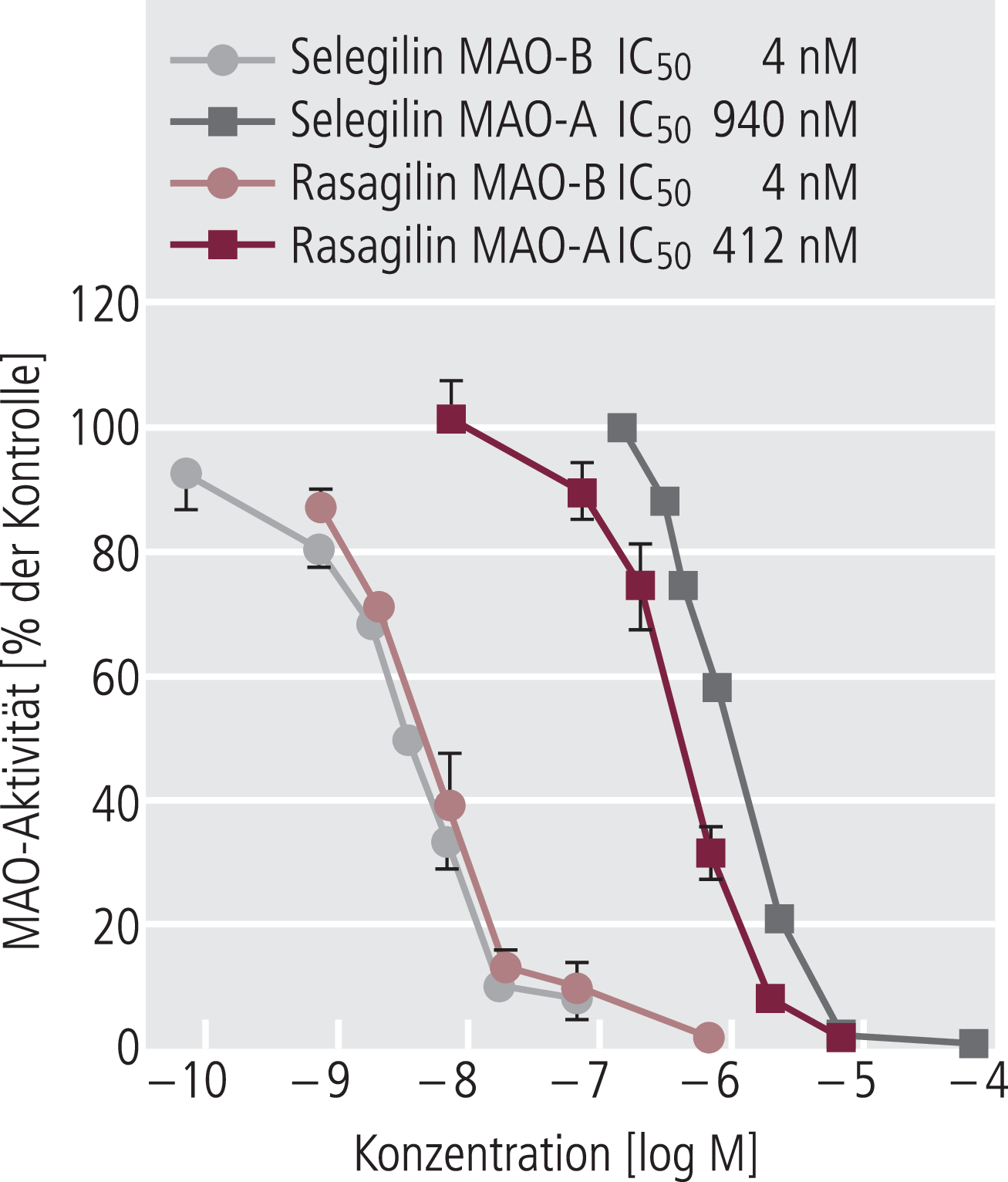
Abb. 2. Wirkstärken von Selegilin und Rasagilin als akute Hemmstoffe der Monoaminoxidasen MAO-A und MAO-B in Hirnhomogenaten der Ratte in vitro; Dosis-Wirkungs-Kurven und halbmaximale Hemmkonzentrationen (IC50) aus Youdim et al. 2001 [71]
Die absolute Potenz von Rasagilin als Hemmstoff der MAO-B ist etwa 100-mal stärker als für MAO-A. Auch bei In-vivo-Gabe bleiben sowohl der Potenzunterschied wie auch die relativen Selektivitäten für MAO-A und -B erhalten. In Abhängigkeit von der Applikationsart können bei Selegilin die In-vivo-Wirkstärken variieren, bedingt durch den ausgeprägten First-Pass-Effekt nach oraler Gabe. Im Hinblick auf die Hemmung von MAO-B sind beide Substanzen in der Anwendung letztlich sehr ähnlich, da die geringe Hemmstärke, aber auch die schlechtere Bioverfügbarkeit von Selegilin durch die deutlich höhere therapeutische Dosis (10 mg/Tag) im Vergleich zu Rasagilin (1 mg/Tag) ausgeglichen wird.
Die relative Selektivität beider Verbindungen für das Monoaminoxidase-B-Isoenzym hat deutliche sicherheitspharmakologische Aspekte, zum einen, weil eine zu starke Hemmung der Monoaminoxidase A mit dem sogenannten „cheese-effect“ assoziiert sein kann, einer akuten Blutdrucksteigerung nach Aufnahme von Tyramin-reicher Nahrung. Im Hinblick auf die experimentell belegte relativ große Selektivität von Selegilin und Rasagilin als Monoaminoxidase-B-Inhibitoren wird in den jeweiligen Fachinformationen für beide Substanzen kein Warnhinweis auf die gleichzeitige Aufnahme Tyramin-haltiger Nahrung gegeben. Dies ist im Prinzip auch in einer aktuellen Studie bestätigt worden [33], in der für 10 mg Selegilin ein sogenannter Tyramin-Sensitivitätsfaktor (TSF) von 2,5 ermittelt wurde (im Vergleich zu 1,5 in der Plazebo-Gruppe). Für die analoge therapeutische Dosierung von Rasagilin (1 mg pro Tag) wurde ein etwas geringerer TSF-Wert von 2,0 ermittelt, der sich bei Verdoppelung der Dosis auf 2,4 nur leicht erhöhte. Dem gegenüber steht eine ältere Publikation von Schulz et al. [62], die für eine Verdoppelung der therapeutischen Dosis von Selegilin von 10 mg auf 20 mg pro Tag ein deutlich höheres Risiko für eine Tyramin-bedingte Blutdrucksteigerung gezeigt hatte. In einer Untersuchung mit Parkinson-Patienten [16] konnten unter der Therapie mit Rasagilin (1 mg, 2 mg; vergleichbar zu 10 mg bzw. 20 mg Selegilin) kein besonderer Effekt einer Tyramin-Gabe auf den Blutdruck gesehen werden. Damit hat möglicherweise Rasagilin in therapeutischen Dosierungen (1 mg bis 2 mg pro Tag) einen leichten Vorteil im Hinblick auf das allgemein aber sehr geringe Risiko eines Tyramin-Effekts.
Zum anderen wurden für Selegilin einige Kasuistiken eines Serotonin-Syndroms auch bei therapeutischer Dosierung beschrieben. Diese sehr seltene, aber potenziell gefährliche Arzneimittelinteraktion wird meist ausgelöst durch die Einnahme von zwei Substanzen, die über unterschiedliche Mechanismen zu einer Erhöhung der zerebralen Serotonin-Konzentration führen können. Die gleichzeitige Einnahme von Selegilin mit anderen Serotonin-verstärkenden Substanzen, beispielsweise selektiven Wiederaufnahmehemmern (SSRI), gilt daher in der Fachinformation als Gegenanzeige [22]. Für Rasagilin ist der Hinweis nur unter Warnhinweisen, nicht aber unter Gegenanzeigen aufgeführt [21], da für Rasagilin keine Daten vorliegen und man die Ähnlichkeit zu Selegilin hier als Basis genommen hat.
Eine kürzlich erschienene experimentelle Arbeit weist nun in die Richtung, dass möglicherweise zwischen beiden Substanzen auch ein grundsätzlicher Unterschied besteht. Neurotoxisch im Sinne einer Serotonin-Verstärkung zu interpretierende Effekte an der Ratte wurden hier für die Kombination des SSRI Fluoxetin mit Selegilin, aber nicht für die Kombination mit Rasagilin beschrieben [64]. Auch in der Arbeit von Finberg und Youdim [26] wurde ein signifikant höherer Verhaltensscore für die Kombination Fluoxetin und Selegilin (10 mg/kg) als für Fluoxetin und Rasagilin (10 mg/kg) gesehen, obwohl die Dosis relativ gesehen für Rasagilin sehr viel höher ist. In aller Vorsicht könnten diese Daten ein weiteres Indiz dafür sein, dass im Hinblick auf diese zwar seltene, aber dann potenziell bedrohliche Arzneimittelinteraktion Rasagilin im Vergleich zu Selegilin einen Vorteil hat. Für beide Substanzen gilt, dass bei Intoxikationen die Selektivität für MAO-B nachlässt und damit das Risiko für ein Serotonin-Syndrom bei entsprechender Komedikation steigt.
Pharmakodynamische Bedeutung der Metaboliten
Psychotomimetische UAW
Während Rasagilin fast ausschließlich zu Aminoindan abgebaut wird, das im Hinblick auf eine MAO-B-Hemmung und auf vasoaktive Eigenschaften im relevanten Konzentrationsbereich nicht wirksam ist [12, 13], gegebenenfalls aber eigenständige neuroprotektive Eigenschaften hat (siehe unten), wird Selegilin außer zum inaktiven Desmethylselegilin zu hoch wirksamem Methamphetamin und Amphetamin abgebaut (Abb. 1). Beide Verbindungen kumulieren bei akuter, vor allem aber bei chronischer Behandlung mit Selegilin zu beträchtlichen Plasmakonzentrationen (Tab. 2) [39], die allerdings nicht so hoch sind, dass man bei Selegilin regelhaft mit psychotomimetischen Amphetamin-artigen UAW rechnen müsste. Im Einzelfall scheinen sie aber ausreichend hoch zu sein, so dass Amphetamin-artige oder besser Amphetamin-ähnliche UAW unter therapeutischen Bedingungen mit Selegilin eine deutliche Rolle spielen [22, 48]. Dies gilt für die Monotherapie mit Selegilin, aber auch für die Kombinationstherapie mit Levodopa. Unter Rasagilin sind diese UAW eher auf Plazebo-Niveau [31, 43]. Amphetamin-ähnliche ZNS-Nebenwirkungen von Selegilin wurden von Nickel et al. [50, 51] in EEG-Untersuchungen an Ratten gesehen. Hier wurden auch Hinweise auf ein mögliches Abhängigkeitspotenzial von Selegilin gefunden.
Kürzlich publizierte Daten zu elektroenzephalographischen Untersuchungen an der wachen Ratte zeigen deutliche Unterschiede zwischen Selegilin auf der einen Seite sowie Rasagilin und 1-Aminoindan auf der anderen Seite und werden im Sinne einer Beteiligung der Amphetamin-Metaboliten im Gesamtwirkungsspektrum von Selegilin interpretiert [17]. Ein weiterer Hinweis Amphetamin-Metabolit-vermittelter Effekte von Selegilin ist eine erhöhte Expression des Dopamin-Transporters (DAT), die von Lamensdorf et al. [40] nach subchronischer Behandlung mit Selegilin, nicht aber mit Rasagilin, im Rattenhirn gesehen wurde. Auch der Befund, dass Selegilin, nicht aber Rasagilin, die Effekte der für zentrale noradrenerge Neurone toxischen Verbindung DSP-4 aufheben kann, wurde über die Amphetamin-artige Wirkung der Selegilin-Metaboliten (Hemmung der neuronalen Aufnahme) erklärt [44].
Schlafstörungen sind eine bekannte und auch plausible Komplikation der Therapie mit Stimulanzien. Daher ist es auch verständlich, dass Schlafprobleme eine der häufigen UAW von Selegilin sind [22]. Distinkte, aber deutliche Effekte einer subchronischen Behandlung mit Selegilin konnten auch schlafpolygraphisch gezeigt werden [67]. Hier kam es besonders zu einer Reduktion von REM-Schlaf und einer Verlängerung der REM-Latenz. Obwohl ein Einfluss der MAO-B-Hemmung auch möglich ist, weisen die Autoren spezifisch darauf hin, dass solche REM-Schlaf-Veränderungen typisch für Amphetamine sind. Schlafprobleme als UAW sind bei Rasagilin sehr selten [21], sowohl in der Monotherapie als auch in Kombination mit anderen Dopaminergika, und waren in einer Sekundäranalyse neuerer klinischer Studien auf Plazebo-Niveau [20].
Kardiovaskuläre UAW
In den ursprünglichen Plazebo-kontrollierten Wirksamkeitsstudien hatte Selegilin sich als sehr gut verträglich in der Therapie des Morbus Parkinson gezeigt. In den folgenden Jahren musste man jedoch einsehen, dass nicht nur psychotomimetische unerwünschte Arzneimittelwirkungen mit relevanten Häufigkeiten auftreten, sondern dass im Einzelfall auch erhebliche kardiovaskuläre UAW mit schweren Blutdruckproblemen und sogar Todesfällen einer Selegilin-Therapie zuzuschreiben waren, wie es in zwei großen Studien beschrieben wurde [7, 42]. Eine kleinere, zum Teil in der Sekundärliteratur überbewertete Studie [60] zeigte diesen Effekt nicht [19]. In der Folgezeit konnte aber, ausgehend von den genannten Beobachtungen, in gezielteren kleineren Untersuchungen eine Störung der Blutdruckregulation im Sinne orthostatischer Blutdrucksenkungen als Folge einer Selegilin-Therapie bei Parkinson-Patienten in Monotherapie, aber auch in Kombination mit anderen dopaminergen Substanzen gesehen werden [14, 15, 58]. Für Rasagilin liegen derartige Meldungen nicht vor [31, 43, 59].
Diese zum Teil schwerwiegenden unerwünschten Arzneimittelwirkungen hat man von Anfang an mit der Bildung von Amphetamin-Metaboliten unter Selegilin-Therapie in Verbindung gebracht, da eine Amphetamin- und Methamphetamin-Gabe zu ähnlichen kardiovaskulären Komplikationen bei gesunden Probanden führen kann. In jüngerer Zeit konnten diese Annahmen durch eine Reihe von klinisch-pharmakologischen und auch tierexperimentellen Arbeiten gut belegt werden (Tab. 3). Zum einen haben schon sehr alte Daten von Martin et al. [47] zeigen, dass eine Amphetamin- und Methamphetamin-Gabe zu ähnlichen kardiovaskulären Komplikationen bei gesunden Probanden führen kann. Zum anderen zeigten Pavese et al. [57] eine Störung der autonomen Blutdruckregulation nach kurzfristiger Methamphetamin-Gabe bei einer Reihe von Parkinson-Patienten im Vergleich zu gesunden Kontrollpersonen. Bei der mechanistischen Erklärung muss man berücksichtigen, dass eine Interferenz von Amphetaminen mit peripheren noradrenergen Mechanismen relativ komplex sein kann, da eine einfache Störung der Noradrenalin-Freisetzung in Abhängigkeit von den relativen Konzentrationen und der Systemempfindlichkeit über prä- oder postsynaptische noradrenerge Rezeptoren sehr unterschiedliche Blutdruckeffekte auslösen kann. Trotzdem konnten verschiedene tierexperimentelle Untersuchungen deutliche, reproduzierbare Blutdruckeffekte von Selegilin bei relevanten Dosierungen zeigen, die in der Regel für Rasagilin in analogen Dosierungen so nicht nachweisbar waren (Tab. 3) [1, 25, 30]. Glezer and Finberg [30] zeigten in Konzentrationen beginnend unter 1 µmol/l spezifische Effekte von Methamphetamin am Vas deferens der Ratte, die auf eine Hemmung der Noradrenalin-Rückaufnahme zurückgehen und die mit der kardiovaskulären unerwünschten Arzneimittelwirkung von Selegilin im Sinne einer Vasodilatation parallel gehen. Besonders empfindlich reagierte hier die präsynaptische Komponente nach elektrischer Feldstimulation auf eine Methamphetamin-Konzentration unter 1 µmol/l. Abassi et al. [1] zeigten ebenfalls an der Ratte, dass bei subchronischer Gabe von Selegilin spezifische Blutdruck-Regulationsstörungen nachweisbar waren, nicht aber bei der Gabe von äquipotenten Rasagilin-Dosen (Tab. 3). Die Effekte und Unterschiede waren zwar nicht sehr stark, aber trotzdem deutlich und signifikant nachweisbar. Noch stärkere Unterschiede zugunsten von Rasagilin wurden beim akuten Vergleich von jeweils 10 mg/kg gesehen, trotz der für Rasagilin ungünstigen, da relativ hohen Dosis (Daten nicht gezeigt). In der klinischen Bewertung dieser Effekte muss man bedenken, dass sie schon an einer relativ kleinen Gruppe von Ratten signifikant nachgewiesen werden konnten, während sie im therapeutischen Bereich mit Selegilin nur als unerwünschte Arzneimittelwirkungen weniger Patienten manifest werden.
Tab. 3. Wichtige Unterschiede zwischen Selegilin und Rasagilin in kardiovaskulär-pharmakologischen Untersuchungen
|
Arbeit |
Modelle |
Substanzen |
Effekt |
|
Glezer and Finberg [30] |
„Twitch response“ des Vas deferens der Ratte nach elektrischer Feldstimulation 1) postsynaptische Komponente (ohne Prazosin) 2) präsynaptische Komponente (mit Prazosin) |
R(–)- und S(+)-Methamphetamin, R(+)-1-Aminoindan, alle 0,1–100 µmol/l |
Hemmung der präsynaptischen α2-Komponente der Twitch Response: Kein Effekt für 1-Aminoindan |
|
Abassi et al. [1] |
Verschiedene kardiovaskuläre Parameter (Blutdruck, Frequenz, Plasmakatecholamine) an der anästhesierten oder wachen Ratte |
21 Tage, oral 10 mg/kg Selegilin, 1 mg/kg Rasagilin |
Über die Therapiedauer von 21 Tagen zum Teil signifikant stärkere Effekte von Selegilin auf systolischen, diastolischen und mittleren arteriellen Druck, nicht aber Frequenz |
|
Finberg et al. [25] |
Verschiedene kardiovaskuläre Parameter an der Ratte bzw. an der dezerebrierten Ratte („pithed“) |
7 Tage oral 1 bzw. 5 mg/kg Selegilin, 0,2 bzw. 1,0 mg/kg Rasagilin, als Einmalgabe i.v. pithed rat |
1. Selegilin, aber nicht Rasagilin verstärkt Hypotension nach Levodopa (7 Tage oral) 2. Selegilin, aber nicht Rasagilin verstärkt Hypotension nach Dopamin (akut, i.v.) 3. Selegilin, aber nicht Rasagilin erhöht Plasmakatecholamine (Abb. 3) |
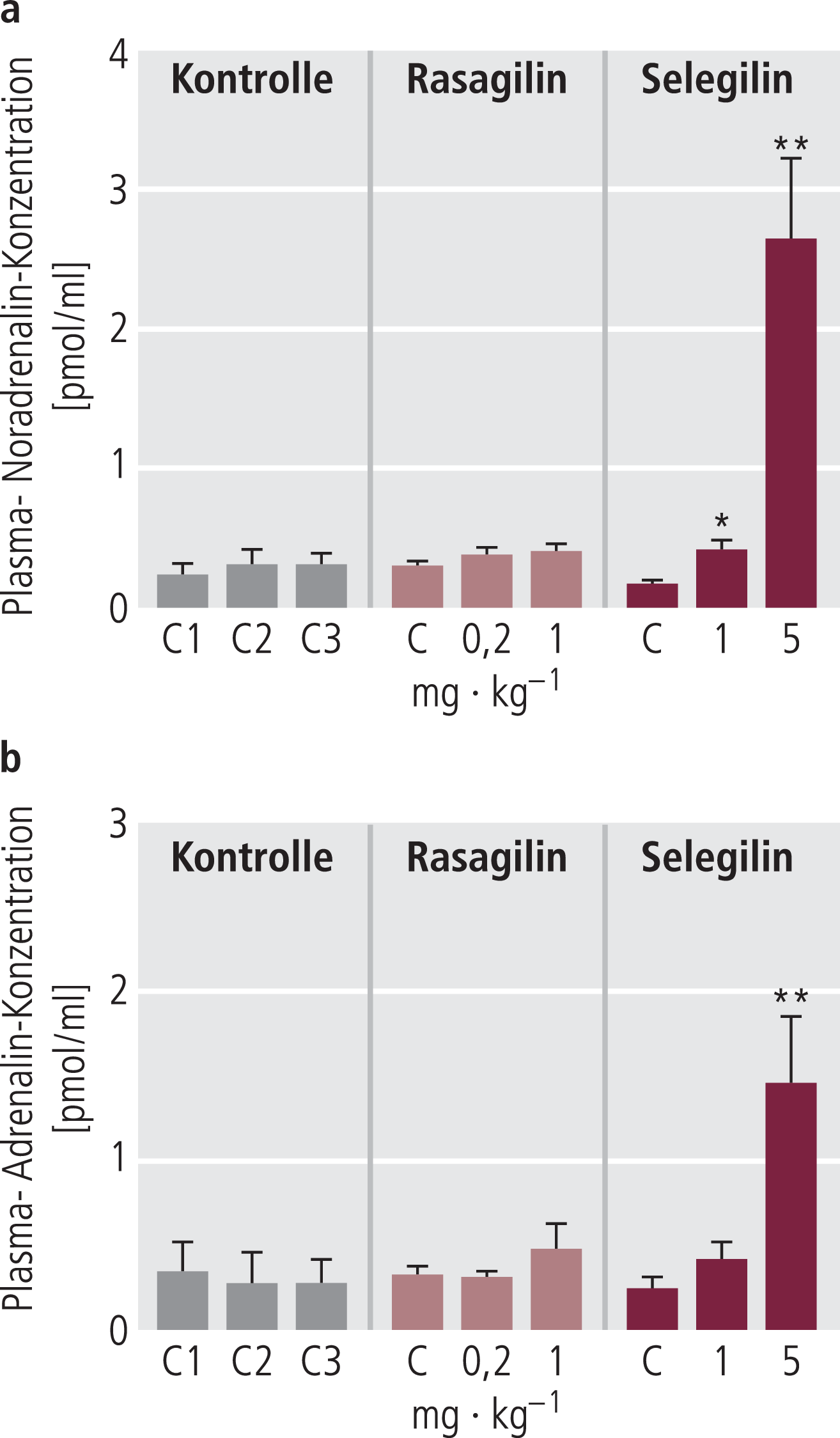
Abb. 3. Plasmakatecholamine an der dezerebrierten Ratte nach Gabe von Selegilin bzw. Rasagilin nach i.v. Bolus. Daten aus Finberg et al., 2006 [25]. C: Kontrolle
Auch in einer weiteren Untersuchung von Finberg et al. [25] konnten kardiovaskuläre Störungen mit Selegilin nach subchronischer Gabe an der Ratte ausgelöst werden, nicht aber mit äquipotenten Dosen von Rasagilin (Tab. 3).
Diese Daten weisen darauf hin, dass auch bei einer Therapie mit Selegilin ausreichende Plasmakonzentrationen der Amphetamin-Verbindungen auftreten können, die distinkte Veränderungen der Blutdruckregulation auslösen können. Sowohl die epidemiologische wie auch die pharmakologische Datenlage zeigen solche Effekte für Rasagilin eher nicht.
Neuroprotektive Eigenschaften
Die positive Wirkung auf die Symptomatik der Parkinson-Erkrankung von Selegilin hat man zunächst ausschließlich mit der durch die Hemmung der Monoaminoxidase B verbundenen Veränderung des Dopamin-Stoffwechsels in Verbindung gebracht. In vielen weiterführenden experimentellen Untersuchungen konnte gezeigt werden, dass Selegilin darüber hinaus neuroprotektive oder neurotrophe Eigenschaften besitzt. Diese sind zum Teil mit der MAO-B-Hemmung assoziiert, werden zum anderen aber unabhängig von der MAO-B-Hemmung vermittelt [35, 49], wobei der Mechanismus nicht geklärt ist. Darüber hinaus muss man berücksichtigen, dass die Amphetamin-Metaboliten neurotoxische Effekte zeigen [2, 4, 10, 38]. Mechanistisch werden hier verstärkter oxidativer Stress und mitochondriale Schädigung durch Amphetamine diskutiert, wie sie häufig bei chronischer Einnahme von Stimulanzien beschrieben werden [9, 29].
Auch für Rasagilin hat man sehr früh schon im Entwicklungsprogramm deutliche neuroprotektive Eigenschaften gesehen, die zum Teil nicht nur deutlich über die bei vergleichbaren Dosen von Selegilin erhaltenen Effekte hinausgehen, sondern unter manchen Bedingungen auch qualitativ neue Aspekte zeigen.
Einen Überblick über relevante Unterschiede zwischen beiden Substanzen in dieser Hinsicht gibt Tabelle 4.
Tab. 4. Wichtige Unterschiede zwischen Selegilin und Rasagilin im Hinblick auf neuroprotektive bzw. neurorestorative Eigenschaften
|
Zitat |
Modell |
Substanzen |
Befund |
|
Finberg et al. [24] Goggi et al. [32] |
Embryonale Rattenhirnzellen in Kultur |
Selegilin 0,1 µmol/l Rasagilin 0,1 µmol/l |
Nur Rasagilin zeigte stimulierte Proliferation nach 6 Tagen Kultur (serumfrei) |
|
Abu-Raya et al. [2] Bar-Am et al. [4] |
PC12-Zellen nach Glucose-Entzug |
Selegilin und Rasagilin (0,01–1,0 µmol/l) |
1. Signifikant stärkere Reduktion von Zelltod für Rasagilin (0,1–1,0 µmol/l) 2. Zelltoxische Effekte von Methamphetamin (0,1–1,0 µmol/l) 3. Signifikante Reduktion von Zelltod durch 1-Aminoindan (1 µmol/l) |
|
Kitani et al. [36] Carrillo et al. [11] |
Ratten |
Rasagilin 0,5 und 1,0 mg/kg Selegilin 0,1; 0,5; 1,0 mg/kg |
Stärkere Expression antioxidativer Enzyme nach Rasagilin; kein direkter Vergleich |
|
Zhu et al. [76] |
C57/BL/6 Mäuse, intrazerebrale Injektion von Lactacystin |
Selegilin 1,0 mg i. p., Rasagilin 0,2 mg i. p. |
Deutlicherer Schutz nigrostriataler dopaminerger Neurone durch Rasagilin |
|
Weinreb et al. [68] |
Ratten |
Selegilin 10 mg/kg für 30 Tage |
Qualitativ und auch quantitativ unterschiedliche Regulation verschiedener Gene im Gehirn mit direkter Relevanz für Regulation von Zellwachstum bzw. Zelltod |
In-vitro-Daten
Einer der ersten Befunde, der auf Unterschiede im neuroprotektiven Potenzial zwischen Selegilin und Rasagilin hinwies, geht auf Finberg et al. [24] zurück. Die Autoren konnten zeigen, dass am Modell embryonaler Rattenhirnzellen in Gewebekultur zwar beide Verbindungen die Anzahl der Tyrosinhydroxylase-sensitiven Neurone erhöhten, aber nur Rasagilin bei Serumentzug neuroprotektiv ist, und zwar in einer Konzentration von 10–7 und 10–6 mol/l. In einer Folgeuntersuchung konnten Goggi et al. [32] dann zeigen, dass der Unterschied zwischen beiden Verbindungen primär bei Serumentzug vorhanden war und nicht in Gegenwart von Serum; aber auch unter Serum-haltigen Bedingungen konnten sie eine konstante etwa um 15 bis 20% bessere neuroprotektive Wirkung von Rasagilin im Vergleich zu Selegilin im gleichen Zellmodell zeigen. Auch unter Benutzung von PC12-Zellen und Glucoseentzug wurden von Abu-Raya et al. [2] deutlich bessere neuroprotektive Eigenschaften für Rasagilin im Vergleich zu Selegilin gezeigt. Interessanterweise hatte in diesem Modell auch der Rasagilin-Metabolit Aminoindan neuroprotektive Eigenschaften, während Methamphetamin, der wichtigste Selegilin-Metabolit, eher neurotoxische Effekte zeigte.
Dieser Befund wurde in einer weiteren Publikation ebenfalls an PC12-Zellen bestätigt, wo auch antiapoptotische Eigenschaften erfasst wurden [4]. Wieder war Rasagilin dem Selegilin überlegen, und positive Effekte waren auch für den Rasagilin-Metaboliten Aminoindan, nicht aber für den Selegilin-Metaboliten Methamphetamin nachweisbar; Letzterer zeigte erneut eher neurotoxische Eigenschaften und verchlechterte die protektiven Eigenschaften von Selegilin (Abb. 4).
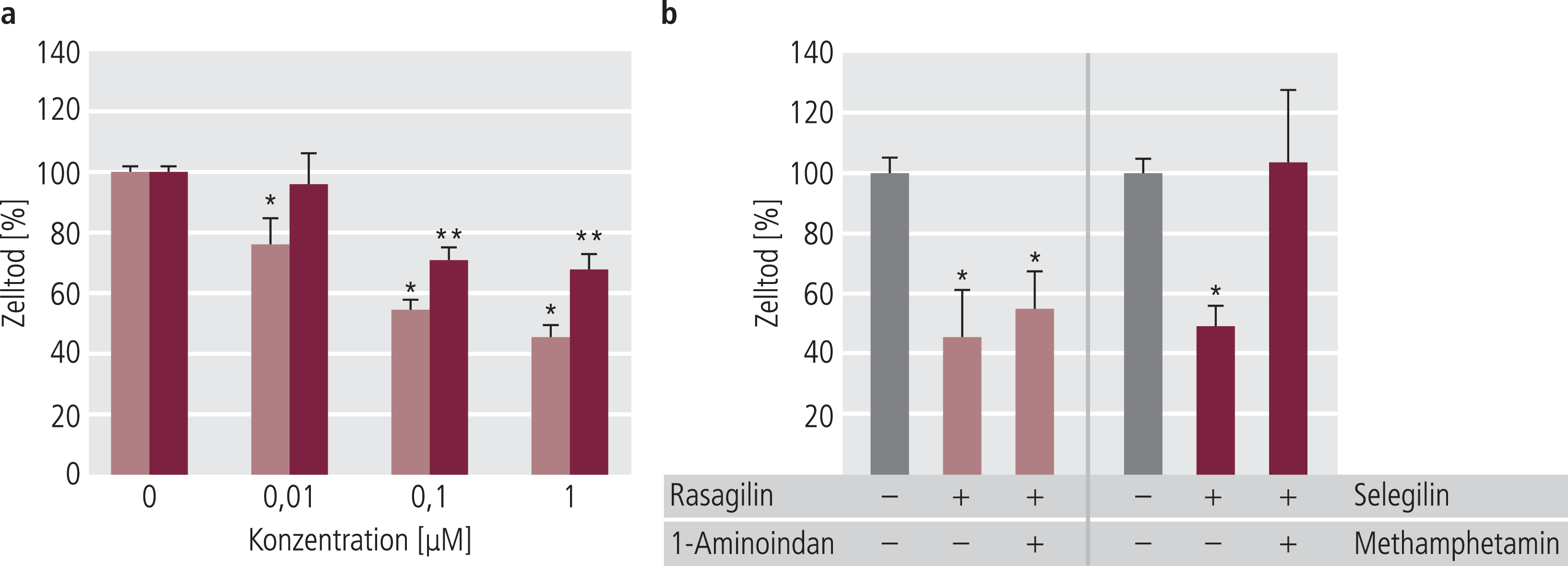
Abb. 4. Neuroprotektive Eigenschaften von Selegilin und Rasagilin an PC12-Zellen nach Glucoseentzug und Sauerstoffentzug im Medium. Gemessen wurde (a) Zelltod über LDH-Freisetzung entweder für beide Substanzen allein oder (b) in Kombination mit Methamphetamin bzw. 1-Aminoindan; alle Substanzen wurden in einer Konzentration von 1 µM eingesetzt. Mod. nach Abu-Raya et al. (2002) [2].
In einer weiteren Arbeit wurden an einer humanen Neuroblastom-Zelllinie (SY5Y) in Gegenwart von Dexamethason zelltoxische Effekte ausgelöst. Diese Effekte konnten durch Selegilin, Rasagilin und 1-Aminoindan reduziert werden [65]. Rasagilin hatte den stärksten Effekt. Die Autoren schließen, dass die überlegenere protektive Wirksamkeit von Rasagilin durch additive Effekte der Muttersubstanz und des Hauptmetaboliten 1-Aminoindan zustande kommt.
In-vivo-Daten
Dass die subchronische Behandlung mit Selegilin zu einer Hochregulation von antioxidativen Enzymen im Rattenhirn führen kann, ist schon seit längerer Zeit bekannt [36]. Auch für Rasagilin konnte von der Gruppe von Kitani dieser Befund belegt werden [11], und erschien deutlich stärker ausgeprägt als bei analogen Untersuchungen mit Selegilin.
Überlegene neuroprotektive Eigenschaften wurden für Rasagilin kürzlich auch in einem Tiermodell der Parkinson-Erkrankung gezeigt. Nach Behandlung mit dem Ubiquitin-Proteasom-Hemmstoff Lactacystin wurden zwar für beide Substanzen neuroprotektive Eigenschaften gesehen, aber nur für Rasagilin wurde eine Rückbildung der nigrostriatalen Degeneration gezeigt, besonders für die Bedingung, wenn beide Substanzen nach Lactacystin-Insult gegeben wurden [76].
Wenn auch der Mechanismus der überlegenen neuroprotektiven Wirkung von Rasagilin nicht sicher aufgeklärt ist, konnten in einer 2009 publizierten Studie [68] auch auf genomischer Ebene Hinweise und Erklärungen für die Unterschiede zwischen beiden Substanzen gefunden werden. Die Autoren untersuchten mithilfe von Microarrays und zweidimensionaler Gelelektrophorese gekoppelt mit Massenspektrometrie Veränderungen der Genexpression im Rattenhirn nach subchronischer Behandlung mit Rasagilin und Selegilin. Neben einer ganzen Reihe von Genen, die durch beide Substanzen gleichgerichtet entweder up- oder down-reguliert wurden, beschreiben die Autoren 10 Gene, die bei der Behandlung der Tiere mit äquipotenten Dosen beider Substanzen unterschiedlich reguliert werden (Tab. 5). Da manche dieser Gene in zellrestaurative Prozesse oder zelltoxische Prozesse eingebunden sind, könnte diese genomische Analyse erste Hinweise auf die mechanistische Erklärung der unterschiedlichen neuroprotektiven Eigenschaften beider Verbindungen geben [68].
Tab. 5. Unterschiedliche Genregulation im Gehirn nach 30 Tagen Behandlung mit Selegilin (10 mg/kg) oder Rasagilin (1 mg/kg) an der Ratte [Daten aus 68]
|
Genprodukt (Protein) |
Selegilin 10 mg/kg |
Rasagilin 1 mg/kg |
|
Aldolase C, Fructosebisphosphate |
D |
NC |
|
Enolase1 protein |
D |
U |
|
Enolase 2, gamma, neuronal |
D |
U |
|
Glutamine synthetase (glutamate-ammonia ligase) |
D |
NC |
|
Transferrin |
D |
NC |
|
Heat shock 70 kDa protein 8 isoform 1 |
D |
NC |
|
Stress-induced-phosphoprotein 1 (Hsp70/Hsp90-organizing protein) |
D |
NC |
|
Phosphatidylethanolamine binding protein, Hippocampal cholinergic neurostimulating peptide |
U |
NC |
|
Guanine nucleotide-binding protein, beta-1 |
D |
NC |
|
Proteasome 26S non-ATPase subunit 9 |
D |
U |
D: Down-Regulation; U: Up-Regulation, NC: Keine Veränderung. Die angegebenen Proteine zeigten für die beiden Behandlungen eine unterschiedliche Genexpression, die auf Proteinebene bestätigt wurde.
Die Rolle von 1-Aminoindan
Ein wichtiger Punkt in der Aufklärung der überlegenen neuroprotektiven Wirkung von Rasagilin liegt in der intrinsischen Wirksamkeit des Hauptmetaboliten 1-Aminoindan, dessen Eigeneffekte immer besser belegt sind [70]. Dies bestätigt auch eine ganz aktuelle Studie [69], wo die Wirksamkeit in zwei In-vivo-Parkinson-Modellen (6-Hydroxydopamin, Lactacystin) gezeigt und bewertet wurde. Einen Gesamtüberblick zu neuroprotektiven In-vitro- und In-vivo-Wirkungen von 1-Aminoindan geben Bar-Am et al. [5], die auch zeigen, dass diese Effekte zum deutlichen Teil unabhängig von der MAO-B-Hemmung und damit von der Analogie zu Selegilin sind. Bestätigt wird die Annahme einer spezifischen Wirkkomponente von 1-Aminoindan in einer aktuellen Studie von Dimpfel und Hoffmann [17], wo die Beeinflussung neuronaler Aktivitätsmuster durch glutamaterge Agonisten von Rasagilin und 1-Aminoindan reduziert werden konnte, nicht aber durch Selegilin.
Neuroprotektion am Menschen
Neuroprotektive Eigenschaften von Selegilin sind schon seit vielen Jahren bekannt und die Diskussion, inwieweit unter chronischer Behandlung von Parkinson-Patienten mit Selegilin auch neuroprotektive Wirkungen bzw. „disease-modifying effects“ vorhanden sind, wurde fast genauso lange heftig geführt. Als Konsens dieser Diskussion wird heute angenommen, dass neuroprotektive Eigenschaften auch am Patienten zwar anzunehmen, aber durch klinische Daten, insbesondere der DATATOP-Studie, nicht belegt sind [3]. Darüber hinaus besteht der Verdacht, dass Methamphetamin-Gebrauch selbst Parkinson auslösen kann [10].
Auch für Rasagilin ist diese Diskussion in den letzten zehn Jahren sehr intensiv geführt worden. Zwei große klinische Studien, die sich besonders auch mit dieser Frage in einem kontrollierten doppelblinden Design beschäftigt haben, sind kürzlich publiziert worden [34, 52, 53, 55, 56]. Die TEMPO-Studie [56, 57] zeigte bei stärker erkrankten Patienten bei einer Dosis von 2 mg/Tag eine Verlangsamung der Progredienz, die im eigentlichen Sinn als „disease-modifying“ interpretiert wurde [34]. Dieser Befund wurde bei den weniger kranken Parkinson-Patienten der ADAGIO-Studie für die 1-mg/Tag-Dosis im Wesentlichen bestätigt [52, 53]. Die Kritik, dass dieser Effekt in der ADAGIO-Studie [52] bei der 2-mg-Dosis nicht signifikant war, könnte auf eine U-förmige Dosis-Wirkungs-Kurve zurückgehen, wie sie auch im Tiermodell für neuroprotektive Effekte von Rasagilin an der Ratte gezeigt wurden [8, 61, 75]. Wenn auch diese beiden großen Studien immer noch einige Fragen offen lassen, scheint sich doch heute die Interpretation durchzusetzen [54], dass neuroprotektive und damit Krankheits-modifizierende Effekte für Rasagilin bei Parkinson-Patienten vorhanden sind [37].
Zusammenfassung
1. Bei Selegilin und Rasagilin handelt es sich um zwei chemisch unterschiedliche irreversible Monoaminoxidase-B(MAO-B)-Inhibitoren mit deutlicher Spezifität, da die Hemmstärke für die MAO-A bei beiden Verbindungen in vitro und in vivo etwa zwei Zehnerpotenzen geringer ist.
2. Selegilin ist ein etwa 5-fach schwächerer Inhibitor von MAO-B als Rasagilin, was allerdings durch eine entsprechend höhere Tagesdosis ausgeglichen wird. Als Konsequenz führen unter therapeutischen Bedingungen beide Substanzen zu einer praktisch 100%igen Hemmung von MAO-B in Gehirn und Thrombozyten.
3. Beide Substanzen haben durch die irreversible Hemmung von MAO-B einen „hit and run“-Wirkungsmechanismus, die biologische Halbwertszeit wird durch die Neusynthese des Enzyms nach Absetzen determiniert. Für beide Substanzen ist nach Absetzen die MAO-B-Aktivität nach 40 Tagen (Gehirn) oder 14 Tagen (Thrombozyten) wieder auf dem Ausgangswert. Für die Steuerung der therapeutischen Wirkung ist daher die klassische Pharmakokinetik weniger relevant. Sie ist für beide Substanzen eher ähnlich mit tmax-Werten von unter einer Stunde und t½-Werten von 1 bis 3 Stunden.
4. Therapeutisch wichtige und relevante pharmakokinetische Unterschiede bestehen zum einen bei der Bioverfügbarkeit, die mit <10% für Selegilin fast schon problematisch gering ist, bei Rasagilin immerhin rund 40% beträgt. Zum anderen ist nur die Pharmakokinetik von Rasagilin im therapeutischen Bereich linear, bei Selegilin aber nicht, so dass die Plasmaspiegel bei Dosiserhöhung überproportional steigen.
5. Während Selegilin sehr stark zu den sympathomimetischen und psychotomimetischen Substanzen Methamphetamin und Amphetamin abgebaut wird, die erhebliche Plasmakonzentrationen erreichen, ist der einzige Metabolit des Rasagilins (1-Aminoindan) in dieser Richtung nicht wirksam.
6. Die Amphetamin-Metaboliten werden für die häufigeren psychotomimetischen UAW des Selegilins und Schlafstörungen verantwortlich gemacht. Darüber hinaus belegen experimentelle Untersuchungen, dass unter Selegilin sehr viel stärker und reproduzierbarer Kreislaufveränderungen nachweisbar sind, die zwar nicht für häufige, aber potenziell kritische kardiovaskuläre UAW (Hypotonie, Orthostase, Schwindel) des Wirkstoffs verantwortlich gemacht werden.
7. Mechanistisch unklar sind leichte Vorteile von Rasagilin im Hinblick auf eine diätetische Tyramin-Reaktion und etwas deutlichere Vorteile im Hinblick auf das Risiko eines Serotonin-Syndroms bei Kombinationen mit serotoninergen Substanzen wie beispielsweise mit SSRI.
8. Beide Substanzen haben neuroprotektive und neurorestorative Eigenschaften, wahrscheinlich – besonders bei Rasagilin – zum Teil unabhängig von der MAO-B-Hemmung. In vielen experimentellen Ansätzen konnte hier eine überlegene Wirksamkeit von Rasagilin gezeigt werden. Dazu trägt wahrscheinlich die neuroprotektive Eigenwirkung von 1-Aminoindan bei, während die Amphetamin-Metaboliten von Selegilin eigenständige neurotoxische Eigenschaften haben und neuroprotektive Effekte von Selegilin reduzieren können.
9. Die überlegenen neuroprotektiven Eigenschaften von Rasagilin bilden sich auch in der klinischen Datenlage ab, wo bis jetzt für Rasagilin wesentlich klarere Evidenzen für eine neuroprotektive, eigentlich „disease modifying“-Wirksamkeit gezeigt werden konnten.
10. Rasagilin zeigt damit signifikante und relevante Vorteile im Hinblick auf die Pharmakokinetik (Linearität, Bioverfügbarkeit) und wirksame Metaboliten (keine psychotomimetischen und neurotoxischen Amphetamine, sondern das eigenständig neuroprotektive Aminoindan). Als Folge sind psychotomimetische und kardiovaskuläre UAW weniger zu erwarten. Die überlegene neuroprotektive Wirkung bildet sich auch in aktuellen klinischen Langzeitstudien ab. Die Einschätzung dieser distinkten, aber doch deutlichen Vorteile von Rasagilin gegenüber der älteren Verbindung Selegilin schlägt sich auch in ganz aktuellen vergleichenden Stellungnahmen zu beiden Substanzen in der US-amerikanischen Literatur nieder [37].
Interessenkonflikte
WEM hat Vortrags- bzw. Beraterhonorare von Bristol-Myers Squibb, Cassellamed, Lundbeck, Novartis, Schwabe, Teva und UCB sowie Forschungsunterstützung von Cassellamed, Lundbeck, Schwabe und UCB erhalten.
HR hat u.a. Vortrags- bzw. Beraterhonorare von TEVA/Lundbeck erhalten.
Literatur
1. Abassi ZA, Binah O, Youdim MB. Cardiovascular activity of rasagiline, a selective and potent inhibitor of mitochondrial monoamine oxidase B: comparison with selegiline. Br J Pharmacol 2004;143:371–8.
2. Abu-Raya S, Tabakman R, Blaugrund E, Trembovler V, et al. Neuroprotective and neurotoxic effects of monoamine oxidase-B inhibitors and derived metabolites under ischemia in PC12 cells. Eur J Pharmacol 2002;434:109–16.
3. Ahlskog JE, Uitti RJ. Rasagiline, Parkinson neuroprotection, and delayed-start trials: still no satisfaction? Neurology 2010;74:1143–8.
4. Bar Am O, Amit T, Youdim MB. Contrasting neuroprotective and neurotoxic actions of respective metabolites of anti-Parkinson drugs rasagiline and selegiline. Neurosci Lett 2004;355:169–72.
5. Bar-Am O, Weinreb O, Amit T, Youdim MB. The neuroprotective mechanism of 1-(R)-aminoindan, the major metabolite of the anti-parkinsonian drug rasagiline. J Neurochem 2010;112:1131–7.
6. Bench CJ, Price GW, Lammertsma AA, Cremer JC, et al. Measurement of human cerebral monoamine oxidase type B (MAO-B) activity with positron emission tomography (PET): a dose ranging study with the reversible inhibitor Ro 19–6327. Eur J Clin Pharmacol 1991;40:169–73.
7. Ben-Shlomo Y, Churchyard A, Head J, Hurwitz B, et al. Investigation by Parkinson’s Disease Research Group of United Kingdom into excess mortality seen with combined levodopa and selegiline treatment in patients with early, mild Parkinson’s disease: further results of randomised trial and confidential inquiry. BMJ 1998;316:1191–6.
8. Blandini F. Neuroprotection by rasagiline: a new therapeutic approach to Parkinson’s disease? CNS Drug Rev 2005;11:183–94.
9. Brown JM, Yamamoto BK. Effects of amphetamines on mitochondrial function: role of free radicals and oxidative stress. Pharmacol Ther 2003;99:45–53.
10. Callaghan RC, Cunningham JK, Sajeev G, Kish SJ. Incidence of Parkinson’s disease among hospital patients with methamphetamine-use disorders. Mov Disord 2010;25:2333–9.
11. Carrillo MC, Minami C, Kitani K, Maruyama W, et al. Enhancing effect of rasagiline on superoxide dismutase and catalase activities in the dopaminergic system in the rat. Life Sci 2000;67:577–85.
12. Chen JJ, Swope DM. Clinical pharmacology of rasagiline: a novel, second-generation propargylamine for the treatment of Parkinson’s disease. J Clin Pharmacol 2005;45: 878–94.
13. Chen JJ, Swope DM, Dashtipour K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin Ther 2007;29:1825–49.
14. Churchyard A, Mathias CJ, Boonkongchuen P, Lees AJ. Autonomic effects of selegiline: possible cardiovascular toxicity in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1997;63:228–34.
15. Churchyard A, Mathias CJ, Lees AJ. Selegiline-induced postural hypotension in Parkinson‘s disease: a longitudinal study on the effects of drug withdrawal. Mov Disord 1999;14:246–51.
16. deMarcaida JA, Schwid SR, White WB, Blindauer K, et al.; Parkinson Study Group TEMPO; PRESTO Tyramine Substudy Investigators and Coordinators. Effects of tyramine administration in Parkinson’s disease patients treated with selective MAO-B inhibitor rasagiline. Mov Disord 2006;21:1716–21.
17. Dimpfel W, Hoffmann JA. Electropharmacograms of rasagiline, its metabolite aminoindan and selegiline in the freely moving rat. Neuropsychobiology 2010;62:213–20.
18. Dimpfel W, Hoffmann JA. Effects of rasagiline, its metabolite aminoindan and selegiline on glutamate receptor mediated signalling in the rat hippocampus slice in vitro. BMC Pharmacol 2011;11:2.
19. Donnan PT, Steinke DT, Stubbings C, Davey PG, et al. Selegiline and mortality in subjects with Parkinson’s disease: a longitudinal community study. Neurology 2000;55:1785–9.
20. Elmer L, Schwid S, Eberly S, Goetz C, et al. Rasagiline-associated motor improvement in PD occurs without worsening of cognitive and behavioral symptoms. J Neurol Sci 2006;248:78–83.
21. Fachinformation Azilect® (Rasagilin), Oktober 2010.
22. Fachinformation Movergan® (Selegilin), Februar 2008.
23. Fernandez HH, Chen JJ. Monoamine oxidase-B inhibition in the treatment of Parkinson’s disease. Pharmacotherapy 2007;27:174S–85S.
24. Finberg JP, Lamensdorf I, Commissiong JW, Youdim MB. Pharmacology and neuroprotective properties of rasagiline. J Neural Transm Suppl 1996;48:95–101.
25. Finberg JP, Gross A, Bar-Am O, Friedman R, et al. Cardiovascular responses to combined treatment with selective monoamine oxidase type B inhibitors and L-DOPA in the rat. Br J Pharmacol 2006;149:647–56.
26. Finberg JP, Youdim MB. Pharmacological properties of the anti-Parkinson drug rasagiline. Neuropharmacology 2002;43:1110–8.
27. Fowler JS, Volkow ND, Logan J, Wang GJ, et al. Slow recovery of human brain MAO B after L-deprenyl (Selegeline) withdrawal. Synapse 1994;18:86–93.
28. Freedman NM, Mishani E, Krausz Y, Weininger J, et al. In vivo measurement of brain monoamine oxidase B occupancy by rasagiline, using (11)C-l-deprenyl and PET. J Nucl Med 2005;46:1618–24.
19. Frey BN, Martins MR, Petronilho FC, Dal-Pizzol F, et al. Increased oxidative stress after repeated amphetamine exposure: possible relevance as a model of mania. Bipolar Disord 2006;8:275–80.
30. Glezer S, Finberg JP. Pharmacological comparison between the actions of methamphetamine and 1-aminoindan stereoisomers on sympathetic nervous function in rat vas deferens. Eur J Pharmacol 2003;472:173–7.
31. Goetz CG, Schwid SR, Eberly SW, Oakes D, et al. Parkinson Study Group TEMPO and PRESTO Investigators. Safety of rasagiline in elderly patients with Parkinson’s disease. Neurology 2006;66:1427–9.
32. Goggi J, Theofilopoulos S, Riaz SS, Jauniaux E, et al. The neuronal survival effects of rasagiline and deprenyl on fetal human and rat ventral mesencephalic neurones in culture. Neuroreport 2000;11:3937–41.
33. Goren T, Adar L, Sasson N, Weiss YM. Clinical pharmacology tyramine challenge study to determine the selectivity of the monoamine oxidase type B (MAO-B) inhibitor rasagiline. J Clin Pharmacol 2010;50:1420–8.
34. Hauser RA, Lew MF, Hurtig HI, Ondo WG, et al.; TEMPO Open-label Study Group. Long-term outcome of early versus delayed rasagiline treatment in early Parkinson’s disease. Mov Disord 2009;24:564–73.
35. Jenner P. Preclinical evidence for neuroprotection with monoamine oxidase-B inhibitors in Parkinson’s disease. Neurology 2004;63(Suppl 2):S13–22.
36. Kitani K, Minami C, Yamamoto T, Maruyama W, et al. Do antioxidant strategies work against aging and age-associated disorders? Propargylamines: a possible antioxidant strategy. Ann N Y Acad Sci 2001;928:248–60.
37. Knudsen-Gerber DS. Selegiline and rasagiline: twins or distant cousins? Guidelines. Consult Pharm 2011;26:48–51.
38. Kupsch A, Sautter J, Götz ME, Breithaupt W, et al. Monoamine oxidase-inhibition and MPTP-induced neurotoxicity in the non-human primate: comparison of rasagiline (TVP 1012) with selegiline. J Neural Transm 2001;108:985–1009.
39. Laine K, Anttila M, Huupponen R, Mäki-Ikola O, et al. Multiple-dose pharmacokinetics of selegiline and desmethylselegiline suggest saturable tissue binding. Clin Neuropharmacol 2000;23:22–7.
40. Lamensdorf I, Porat S, Simantov R, Finberg JP. Effect of low-dose treatment with selegiline on dopamine transporter (DAT) expression and amphetamine-induced dopamine release in vivo. Br J Pharmacol 1999;126: 997–1002.
41. Lee DH, Mendoza M, Dvorozniak MT, Chung E, et al. Platelet monoamine oxidase in Parkinson patients: effect of L-deprenyl therapy. J Neural Transm Park Dis Dement Sect 1989;1:189–94.
42. Lees AJ. Comparison of therapeutic effects and mortality data of levodopa and levodopa combined with selegiline in patients with early, mild Parkinson’s disease. Parkinson’s Disease Research Group of the United Kingdom. BMJ 1995;311:1602–7.
43. Lew MF, Hauser RA, Hurtig HI, Ondo WG, et al. Long-term efficacy of rasagiline in early Parkinson’s disease. Int J Neurosci 2010;120:404–8.
44. Magyar K, Pálfi M, Tábi T, Kalász H, et al. Pharmacological aspects of (–)-deprenyl. Curr Med Chem 2004;11:2017–31.
45. Mahmood I, Marinac JS, Willsie S, Mason WD. Pharmacokinetics and relative bioavailability of selegiline in healthy volunteers. Biopharm Drug Dispos 1995;16:535–45.
46. Mahmood I. Clinical pharmacokinetics and pharmacodynamics of selegiline. An update. Clin Pharmacokinet 1997;33:91–102.
47. Martin WR, Sloan JW, Sapira JD, Jasinski DR. Physiologic, subjective, and behavioral effects of amphetamine, methamphetamine, ephedrine, phenmetrazine, and methylphenidate in man. Clin Pharmacol Ther 1971;12:245–58.
48. Montastruc JL, Chaumerliac C, Desboeuf K, Manika M, et al. Adverse drug reactions to selegiline: a review of the French pharmacovigilance database. Clin Neuropharmacol 2000;23:271–5.
49. Naoi M, Maruyama W. Functional mechanism of neuroprotection by inhibitors of type B monoamine oxidase in Parkinson’s disease. Expert Rev Neurother 2009;9:1233–50.
50. Nickel B, Schulze G, Szelenyi I. Effect of enantiomers of deprenyl (selegiline) and amphetamine on physical abuse liability and cortical electrical activity in rats. Neuropharmacology 1990;29:983–92.
51. Nickel B, Szelenyi I, Schulze G. Evaluation of physical dependence liability of l-deprenyl (selegiline) in animals. Clin Pharmacol Ther 1994;56:757–67.
52. Olanow CW, Hauser RA, Jankovic J, Langston W, et al. A randomized, double-blind, placebo-controlled, delayed start study to assess rasagiline as a disease modifying therapy in Parkinson’s disease (the ADAGIO study): rationale, design, and baseline characteristics. Mov Disord 2008;23:2194–201.
53. Olanow CW, Rascol O, Hauser R, Feigin PD, et al.; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009;361:1268–78.
54. Olanow CW, Rascol O. The delayed-start study in Parkinson disease: can’t satisfy everyone. Neurology 2010;74:1149–50.
55. Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study. Arch Neurol 2002;59:1937–43.
56. Parkinson Study Group. A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol 2004;61:561–6.
57. Pavese N, Rimoldi O, Gerhard A, Brooks DJ, et al. Cardiovascular effects of methamphetamine in Parkinson’s disease patients. Mov Disord 2004;19:298–303.
58. Pursiainen V, Korpelainen TJ, Haapaniemi HT, Sotaniemi AK, et al. Selegiline and blood pressure in patients with Parkinson’s disease. Acta Neurol Scand 2007;115:104–8.
59. Rascol O. Rasagiline in the pharmacotherapy of Parkinson’s disease – review. Expert Opin Pharmacother 2005;6:2061–75.
60. Riederer P, Lachenmayer L, Laux G. Clinical applications of MAO-inhibitors. Curr Med Chem 2004;11:2033–43.
61. Sagi Y, Mandel S, Amit T, Youdim MB. Activation of tyrosine kinase receptor signaling pathway by rasagiline facilitates neurorescue and restoration of nigrostriatal dopamine neurons in post-MPTP-induced parkinsonism. Neurobiol Dis 2007;25:35–44.
62. Schulz R, Antonin KH, Hoffmann E, Jedrychowski M, et al. Tyramine kinetics and pressor sensitivity during monoamine oxidase inhibition by selegiline. Clin Pharmacol Ther 1989;46:528–36.
63. Simpson GM, Frederickson E, Palmer R, Pi E, et al. Platelet monoamine oxidase inhibition by deprenyl and tranylcypromine: implications for clinical use. Biol Psychiatry 1985;20:684–7.
64. Speiser Z, Fine T, Litinetsky L, Eliash S, et al. Differential behavioral syndrome evoked in the rats after multiple doses of SSRI fluoxetine with selective MAO inhibitors rasagiline or selegiline. J Neural Transm 2008;115: 107–16.
65 Tazik S, Johnson S, Lu D, Johnson C, et al. Comparative neuroprotective effects of rasagiline and aminoindan with selegiline on dexamethasone-induced brain cell apoptosis. Neurotox Res 2009;15:284–90.
66. Thébault JJ, Guillaume M, Levy R. Tolerability, safety, pharmacodynamics, and pharmacokinetics of rasagiline: a potent, selective, and irreversible monoamine oxidase type B inhibitor. Pharmacotherapy 2004;24:1295–305.
67. Thornton C, Doré CJ, Elsworth JD, Herbert M, et al. The effect of deprenyl, a selective monoamine oxidase B inhibitor, on sleep and mood in man. Psychopharmacology (Berl) 1980;70:163–6.
68. Weinreb O, Amit T, Sagi Y, Drigues N, et al. Genomic and proteomic study to survey the mechanism of action of the anti-Parkinson’s disease drug, rasagiline compared with selegiline, in the rat midbrain. J Neural Transm 2009;116:1457–72.
69. Weinreb O, Bar-Am O, Prosolovich K, Amit T, et al. Does 1-(R)-aminoindan possess neuroprotective properties against experimental Parkinson‘s disease? Antioxid Redox Signal 2011;14:767–75.
70. Weinreb O, Amit T, Bar-Am O, Youdim MB. Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog Neurobiol 2010;92:330–44.
71. Youdim MB, Gross A, Finberg JP. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol 2001; 132:500–6.
72. Youdim MB, Tipton KF. Rat striatal monoamine oxidase-B inhibition by l-deprenyl and rasagiline: its relationship to 2-phenylethylamine-induced stereotypy and Parkinson’s disease. Parkinsonism Relat Disord 2002;8:247–53.
73. Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 2006;7:295–309.
74. Youdim MB, Bakhle YS. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol 2006;147(Suppl 1):S287–96.
75. Youdim MB. Rasagiline in Parkinson’s disease. N Engl J Med 2010;362:657–58.
76. Zhu W, Xie W, Pan T, Jankovic J, et al. Comparison of neuroprotective and neurorestorative capabilities of rasagiline and selegiline against lactacystin-induced nigrostriatal dopaminergic degeneration. J Neurochem 2008;105:1970–8.
Prof. Dr. Walter E. Müller, Pharmakologisches Institut, Biozentrum der Universität Frankfurt/Main, Campus Riedberg, Max-von-Laue-Straße 9, 60438 Frankfurt/Main, E-Mail: pharmacolnat@em.uni-frankfurt.de
Prof. Dr. Heinz Reichmann, Klinik und Poliklinik für Neurologie, Universitätsklinikum Carl Gustav Carus, Technische Universität Dresden, Fetscherstraße 74, 01307 Dresden, E-Mail: Heinz.Reichmann@uniklinikum-dresden.de
Pharmacokinetic and pharmacodynamic properties of selegiline and rasagiline
Selegiline and rasagiline are irreversible inhibitors of monoamine oxidase type B (MAO-B). Due to the weaker potency and the lower bioavailability of selegiline its daily dose is about ten times higher to give the same level of MAO-B inhibition. Both drugs have rather short elimination half-lives. However, due to the irreversible binding to MAO-B, both block MAO-B in human brain for about 40 days. Selegiline is metabolized to a large extent to amphetamine and methamphetamine which are relevant for the side effect profile, rasagiline is metabolized to 1-aminoindan, which contributes to the neuroprotektive properties. The amphetamine metabolites very likely contribute to more common neurological (e.g. sleep disturbances) and cardiovascular (e.g. tachycardia, hypotension) side effects of selegiline relative to rasagiline. Both drugs show neuroprotective effects which are more pronounced for rasagiline than for selegiline, probably because of the rasagiline metabolite 1-aminoindan. The latter probably explains stronger disease-modifiing effects of rasagiline relative to selegiline when given to Parkinson patients.
Key words: Selegiline, rasagiline, pharmacokinetics, pharmacodynamic, differences
Psychopharmakotherapie 2012; 19(05)