Gökce Orhan, Tübingen, Thomas V. Wuttke, Boston/Cambridge, Anne T. Nies, Matthias Schwab, Stuttgart/Tübingen, und Holger Lerche, Tübingen
*In den USA ist die Wirkstoffbezeichnung „ezogabine“ gebräuchlich. Es wurde am 10. Juni 2011 unter dem Handelsnamen PotigaTM zugelassen.
Epilepsie ist eine der häufigsten chronischen Krankheiten des zentralen Nervensystems und betrifft etwa 1% der Weltbevölkerung, entsprechend 800000 Menschen in Deutschland. Die Krankheit ist durch unterschiedliche Arten epileptischer Anfälle charakterisiert, die durch abnorme, exzessive und synchrone neuronale Entladungen im Gehirn ausgelöst werden. Trotz wissenschaftlicher Fortschritte in der Erforschung und im Verständnis der Pathophysiologie von Anfällen und trotz der Entwicklung zahlreicher (>20) Arzneistoffe mit verschiedenen Wirkungsmechanismen, die Anfälle unterdrücken (Antikonvulsiva oder Antiepileptika, im Folgenden als AED=Antiepileptic drug abgekürzt), gibt es immer noch etwa 30% therapierefraktäre Fälle, die nicht auf die bisher verfügbaren Substanzen ansprechen [5, 24]. Dieser hohe Anteil an pharmakoresistenten Epilepsiepatienten belegt den Bedarf an der Entwicklung neuer Medikamente mit vornehmlich neuen Wirkprinzipien in der Hoffnung, dass jede neue Entwicklung eine besondere Chance für therapierefraktäre Patienten bietet.
Retigabin (Abb. 1), der Wirkstoff in Trobalt®*, ist eine interessante Substanz, weil es einen neuen Wirkungsmechanismus aufweist, der von keinem der verfügbaren AED abgedeckt wird. Es verstärkt die Aktivität neuronaler spannungsabhängiger Kaliumkanäle der KV7-Familie (KV7.2–7.5), die von den Genen KCNQ2–5 kodiert werden. Neuronale KV7-Kanäle modulieren die Erregbarkeit von sehr vielen verschiedenen Typen zentraler und peripherer Nervenzellen, indem sie das Membranpotenzial im Subschwellenbereich eines Aktionspotenzials regulieren [7]. Mutationen in neuronalen KV7-Kanälen verursachen erbliche Formen der Epilepsie und der Übererregbarkeit peripherer Nerven [27]. Diese Arbeit fasst die wichtigsten publizierten pharmakologischen und klinischen Daten des neuen Kaliumkanalöffners Retigabin zusammen.
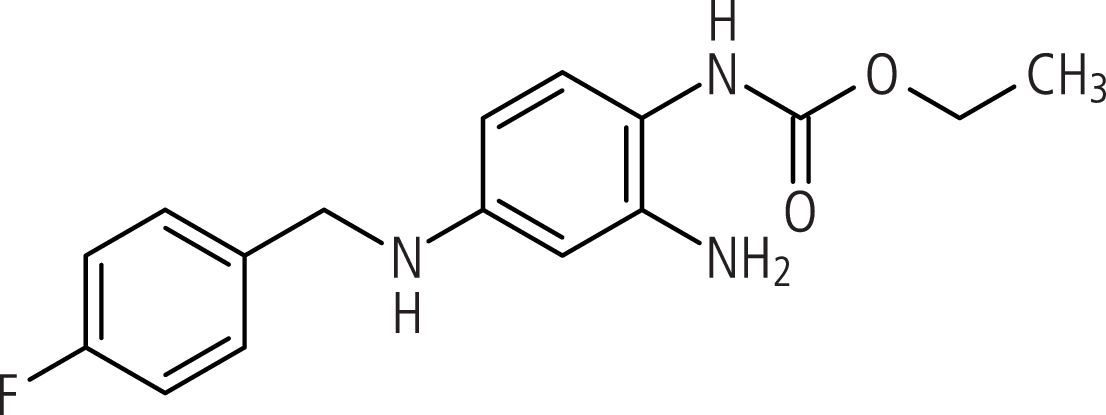
Abb. 1. Retigabin
Arzneistoffsteckbrief
|
Arzneistoffname |
Retigabin (RGB), in den USA Ezogabin (EGB) |
|
Indikation |
Add-on-Therapie für therapierefraktäre fokale Epilepsien |
|
Darreichungsform |
p.o. als Filmtablette |
|
Chemische Struktur |
Abb. 1 |
|
Klinische Studien |
Multizentrische, randomisierte, Plazebo-kontrollierte Doppelblindstudien bei Erwachsenen mit ≥4 fokalen Anfällen pro Monat trotz antikonvulsiver Therapie mit maximal 3 konkurrierenden Antiepileptika. (Phase-II-Dosisfindungsstudie und zwei Phase-III-Studien [RESTORE 1 und 2]) |
|
Nebenwirkungen (UAW) [nach Fachinformation Trobalt®] |
Dosisabhängige, hauptsächlich ZNS-generierte Nebenwirkungen Sehr häufige UAW (>10%): Schwindel, Müdigkeit und Erschöpfung Häufige UAW (1–10%): Amnesie, Aphasie, Koordinationsstörungen, Drehschwindel, Parästhesien, Tremor, Gleichgewichtsstörungen, Gedächtnisstörungen, Wortfindungs- und Verständnisstörungen, Dysarthrie, Aufmerksamkeitsstörungen, Gangstörungen, Myoklonien, Verwirrtheitszustände, psychotische Störungen, Halluzinationen, Desorientierung, Angst, Gewichtszunahme, Appetitzunahme Diplopie, Verschwommensehen, Übelkeit, Obstipation, Dyspepsie, Mundtrockenheit, erhöhte Leberwerte, Dysurie, Harnentleerungsverzögerung, Hämaturie, Chromaturie, Asthenie, Unwohlsein, periphere Ödeme. Gelegentliche UAW (0,1–1%): Hypokinesie, Schluckstörungen, Hautausschlag, Hyperhidrosis, Harnverhalt, Nierensteine |
Präklinische Entwicklung
Retigabin (ATC-Code: N03AX21) ist ein Carbaminsäurederivat (N-(2-Amino-4-(4-fluorbenzylamino)-phenyl)carbaminsäureethylester). Es ist chemisch verwandt mit Flupirtin, einem zentralwirksamen Analgetikum. Ausgehend von Flupirtin wurden mehrere Substanzen synthetisiert, unter denen Retigabin durch seine überlegene antikonvulsive Wirkung auffiel. Retigabin wurde daraufhin in einer Vielzahl von In-vivo- und In-vitro-Modellen getestet und unterdrückte epileptische Aktivität unter anderem in elektrischen [41] und chemischen [1, 34] Anfallsmodellen, im Amygdala-Kindling-Modell [41] sowie dem Niedrig-Mg2+- [2, 8] und Niedrig-Ca2+-Modell an Rattenhirnschnitten [8]. Darüber hinaus zeigte Retigabin Wirksamkeit gegen audiogene Anfälle in DBA/2-Mäusen [34] sowie gegen epileptische Aktivität in humanen Hirnschnitten pharmakoresistenter Patienten [38].
Wirkungsmechanismus und Bedeutung von Kv7-Kanälen
Erste In-vitro-Experimente zur Aufklärung des Wirkungsmechanismus von Retigabin zeigten, dass Retigabin einen K+-Strom in kortikalen Neuronen und mit Nervenwachstumsfaktor (NGF) behandelten PC12-Zellen (Phäochromozytom-Zelllinie) induziert [36]. Nachfolgende Studien demonstrierten eine Interaktion von Retigabin mit in CHO(Chinese hamster ovary)-Zellen überexprimierten KV7.2/KV7.3-Kanälen. Eine genaue Analyse des spannungsabhängigen Schaltverhaltens und der Vergleich mit den zuvor beschriebenen Retigabin-aktivierten Strömen in kortikalen Neuronen und PC12-Zellen legten nahe, dass Retigabin spezifisch Kanäle der KV7-Kaliumkanalfamilie aktiviert [35]. Als Wirkungsmechanismus konnte schließlich eine Retigabin-induzierte Verschiebung der Aktivierungskurve von KV7.2/KV7.3-Kanälen in hyperpolarisierender Richtung identifiziert werden [26, 35, 40, 42], die zu einer vermehrten Öffnung von KV7.2/KV7.3-Kanälen im Subschwellenbereich eines Aktionspotenzials führt.
KV7-Kanäle vermitteln den sogenannten M-Strom, einen langsam aktivierenden und nicht inaktivierenden K+-Strom, dem eine hohe Bedeutung bei der Feinregulierung der neuronalen Feuerungsrate zukommt, weil die spannungsabhängige Offenwahrscheinlichkeit im Subschwellenbereich stark ansteigt [7]. Über die Jahre hinweg wurde eine Vielzahl von Krankheits-assoziierten Mutationen in KV7.1–4 beschrieben, die durch einen Funktionsverlust zu kardialer oder neuronaler Hyperexzitabilität führen und das pathophysiologische Korrelat mehrerer erblicher Erkrankungen wie des Long-QT-Syndroms Typ 1 (LQTS1) (KV7.1), benigner familiärer neonataler Anfälle (BFNS) (KV7.2 und KV7.3) und progressiver Taubheit (KV7.4) [22, 27] darstellen. Die Aktivierung des M-Stroms durch Retigabin führt zu einer Hyperpolarisation der neuronalen Zellmembran und wirkt somit epileptischer Aktivität entgegen. Von großer Bedeutung für das Risikoprofil von Retigabin – vor allem im Hinblick auf kardiale Arrhythmien – ist, dass es ausschließlich KV7.2–5, aber nicht den kardialen KV7.1-Kanal aktiviert [22, 35, 37, 42–44].
Arbeiten unserer und einer anderen Arbeitsgruppe haben in den vergangenen Jahren mehrere Aminosäuren in KV7.2/KV7.3 beschrieben, die essenziell für die Interaktion von Retigabin mit KV7-Kanälen sind. Basierend auf unseren Ergebnissen und der Kristallstruktur zweier bakterieller Kaliumkanäle konnte ein Modell für die Bindungsstelle von Retigabin im Bereich der Kanalpore (S5/S6-Segmente und H5-Porenhelix) von KV7.2/KV7.3 entwickelt werden, das eine Stabilisierung des offenen Zustands des Kanals durch Bindung von Retigabin in der Porenregion nahelegt [25, 37, 44].
Als Kaliumkanalöffner repräsentiert Retigabin eine neue Klasse von AED, mit der die Hoffnung verbunden ist, Patienten erfolgreich zu behandeln, deren epileptische Anfälle mit den aktuell auf dem Markt verfügbaren Antikonvulsiva nicht zufriedenstellend kontrollierbar sind. Eine weitere Verbesserung, auch in Hinblick auf eine größere therapeutische Breite und weniger Nebenwirkungen, ist eventuell durch die Entwicklung von Derivaten mit erhöhter Subklassenspezifität (z.B. ausschließliche Aktivierung von KV7.2 und/oder KV7.3) denkbar.
Neben der Aktivierung von KV7-Kanälen führt Retigabin in sehr viel höheren Konzentrationen unter anderem zu einer verstärkten GABAergen Inhibition [30] und zu einer unspezifischen Blockade von Na+-, Ca2+- und Kainat-induzierten Strömen [35]. Dies scheint allerdings in den beim Menschen angewendeten (und zugelassenen) Dosierungen kaum von klinischer Bedeutung zu sein.
Pharmakokinetik und Metabolismus
Nach oraler Applikation weist Retigabin eine absolute Bioverfügbarkeit von ~60% auf [20]. Retigabin wird schnell resorbiert, und die Plasmaspitzenkonzentration (Cmax) wird typischerweise nach 30 bis 120 Minuten erreicht [9, 10, 15, 19]. Nach einer fettreichen Mahlzeit ist Cmax mäßig erhöht, die Fläche unter der Plasmakonzentrations-Zeit-Kurve (AUC) bleibt hingegen unverändert [28]. Retigabin wird nach einer Kinetik 1. Ordnung eliminiert, wobei die mittlere terminale Halbwertszeit etwa 7,5 Stunden (6,7–8,5 Stunden) ist und die Clearance 0,58 bis 0,76 l·h–1·kg–1 beträgt [9, 10, 19]. Bei jungen Frauen können (aufgrund von Unterschieden im Körpergewicht) bis zu 56% höhere Cmax-Werte als bei Männern erreicht werden, die Clearance ist allerdings vergleichbar [19]. Retigabin hat ein großes Verteilungsvolumen von 8,7 l·kg-1, seine Plasmaproteinbindung beträgt 60 bis 80% [10, 28]. Die Clearance und das Verteilungsvolumen scheinen einer ethnischen Variabilität zu unterliegen und waren bei schwarzen Amerikanern um 25% bzw. 30% geringer als bei kaukasischen Amerikanern [10].
Bei gesunden Freiwilligen konnte gezeigt werden, dass eine lineare Dosis-Wirkungs-Beziehung für Dosierungen von 100 bis 700 mg/Tag besteht, unabhängig davon, ob Retigabin als Einzeldosis oder Mehrfachdosis gegeben wurde [10]. Auch bei Epilepsiepatienten wird Retigabin schnell resorbiert und mit einer terminalen Halbwertszeit von 6 bis 10 Stunden eliminiert. Ebenso wurde eine lineare Dosis-Wirkungs-Beziehung für die therapeutisch eingesetzte Dosierung von 200 bis 400 mg dreimal täglich nachgewiesen [11, 15].
Retigabin wird nicht über Cytochrom-P450(CYP)-Enzyme metabolisiert [17, 29]. Stattdessen erfolgt die Verstoffwechslung über N-Glucuronidierung und in geringerem Maße über N-Acetylierung, wobei der mono-acetylierte Metabolit AWD21-360 entsteht, der ebenfalls glucuronidiert wird [17, 29]. In präklinischen Modellen sind diese Metabolite inaktiv bzw. schwach aktiv [15]. Die Pharmakokinetik von AWD21-360 und Retigabin ist vergleichbar [10, 20]. Durch In-vitro-Studien mit rekombinanten humanen UDP-Glucuronosyltransferasen (UGT) konnte gezeigt werden, dass Retigabin hauptsächlich durch UGT1A1, 1A9, 1A4 und 1A3 glucuronidiert wird [4, 21]. Bei Patienten mit einer verminderten metabolischen UGT1A1-Kapazität (Gilbert-Meulengracht-Syndrom), ist die Pharmakokinetik von Retigabin unverändert, da die fehlende Glucuronidierung durch UGT1A1 durch andere UGT1A-Isoformen kompensiert sowie der mono-acetylierte Metabolit AWD21-360 vermehrt gebildet wird [18]. Andere UGT1- und UGT2-Enzyme scheinen ebenfalls beteiligt zu sein, da trotz eines völligen Verlusts der metabolischen UGT1A-Kapazität (Crigler-Najjar-Syndrom) eine Glucuronidierung von Retigabin in vitro beobachtet werden konnte [4]. Probanden, bei denen die N-Acetylierung aufgrund genetischer Polymorphismen im N-Acetyltransferase-2(NAT2)-Gen verzögert ist („langsame Acetylierer“), hatten eine geringere Exposition zu AWD21-360 als Kontrollprobanden, während die Pharmakokinetik von Retigabin in beiden Gruppen vergleichbar war [18]. Die Verträglichkeit von Retigabin scheint bei Patienten mit Gilbert-Meulengracht-Syndrom oder bei „langsamen Acetylierern“ nicht beeinträchtigt zu sein [18].
Die Ausscheidung von Retigabin und seinen Metaboliten erfolgt fast ausschließlich über die Niere [17, 18]. Eine altersabhängige Verminderung der Retigabin-Clearance von etwa 30% steht im Einklang mit der altersbedingten Verminderung der Nierenfunktion [19]. Die systemische Retigabin-Clearance kann bei Patienten mit mittelschwer oder schwer eingeschränkter Leberfunktion (Child-Pugh-Score ≥7) oder Nierenfunktionsstörung (Creatinin-Clearance <50 bis 80 ml·min–1) bis zu 50% vermindert sein [28]. Deshalb wird eine Dosisanpassung empfohlen [Fachinformation Trobalt®].
Arzneimittelinteraktionen
In-vitro-Studien mit Lebermikrosomen zeigten keine Hemmung von CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 und CYP4A9/11 und eine nur mäßige Hemmung von CYP2A6 durch klinisch relevante Retigabin-Konzentrationen [28]. Darüber hinaus konnte in vitro keine Interaktion von Retigabin und Arzneimitteln, die ebenfalls überwiegend durch Glucuronidierung metabolisiert werden (z.B. Imipramin, Lamotrigin, Valproinsäure, Propofol), nachgewiesen werden [4].
In Studien mit gesunden Freiwilligen wurde keine pharmakokinetische Interaktion zwischen Mehrfachdosen von Retigabin (600 mg/Tag für 3 Tage) und einer Einzeldosis von Phenobarbital (90 mg) beobachtet [9]. Andererseits führten Mehrfachdosen von Phenobarbital (90 mg für 3 Wochen) zu einer 10%igen Erhöhung der AUC von Retigabin, wenn dieses als 200-mg-Einzeldosis gegeben wurde [12]. Die gleichzeitige Gabe von Retigabin und Lamotrigin bei gesunden Freiwilligen führte zu einer wechselseitigen Reduzierung der AUC von 15%, was die Zulassungsbehörden zwar als nicht klinisch relevant angesehen haben, sich aber bei einzelnen Patienten, die einen bestimmten Serumspiegel von Lamotrigin benötigen, kritisch auswirken könnte und deshalb beachtet werden sollte. Mehrfachdosen von Retigabin erhöhten die Lamotrigin-Clearance und verminderten die terminale Halbwertszeit von Lamotrigin [15, 20].
Pharmakokinetische Interaktionen zwischen Retigabin und anderen AED wurden auch in einer Phase-II-Studie mit Epilepsiepatienten untersucht [11, 12]. Es zeigte sich, dass die Pharmakokinetik von Retigabin durch Valproinsäure oder Topiramat nicht beeinflusst wird. Allerdings war die Retigabin-Clearance mäßig erhöht (~30%), wenn Retigabin gleichzeitig mit Phenytoin oder Carbamazepin gegeben wurde. Eine abschließende Beurteilung der klinischen Relevanz dieses Effekts steht wie bei Lamotrigin aus und hängt von den weiteren klinischen Erfahrungen ab.
Im Gegensatz dazu zeigte eine populationspharmakokinetische Auswertung, die klinische Studiendaten von mehr als 800 Patienten einschloss (gepoolte Analyse der Zulassungsstudien), keine klinisch relevanten Effekte von Carbamazepin, Phenytoin oder Phenobarbital auf die Pharmakokinetik von Retigabin [28]. Ebenso hatte Retigabin keinen Einfluss auf die Pharmakokinetik von Phenytoin, Carbamazepin, Valproinsäure oder Topiramat sowie von oralen Kontrazeptiva, die Ethinylestradiol und Norgestrel enthalten [28]. Die Auswertung von Phase-III-Studien zeigte ebenfalls keine klinisch relevanten Effekte von Retigabin auf die Konzentration begleitend gegebener AED, mit Ausnahme eines geringfügigen Abfalls der Lamotrigin-Konzentration, der als nicht klinisch bedeutend eingestuft wurde (aber s.o. Anmerkung der Verfasser) [28]. Kovariate mit signifikanten Effekten auf die Pharmakokinetik von Retigabin sind die Körperoberfläche, das Alter und die Creatinin-Clearance [28].
Klinische Daten
Retigabin bei therapierefraktären fokalen Epilepsien im Erwachsenenalter
Retigabin erwies sich in den bisherigen klinischen und präklinischen Studien als wirksames Add-on-Antiepileptikum bei therapierefraktären fokalen Epilepsien mit oder ohne sekundär generalisierte Anfälle im Erwachsenenalter. Drei multizentrische, randomisierte, Plazebo-kontrollierte Doppelblindstudien bei Erwachsenen mit ≥4 fokalen Anfällen pro Monat trotz antikonvulsiver Therapie mit 1 bis 3 gleichzeitig gegebenen AED (siehe Tab. 1) zeigten eine dosisabhängige Reduktion monatlicher Anfallsfrequenzen und einen dosisabhängigen Anstieg der Responderraten im Vergleich zu Plazebo. Die Ergebnisse dieser drei Studien, eine in der Phase II und zwei in der Phase III, sind alle mittlerweile publiziert [6, 13, 32] und werden im Folgenden zusammengefasst:
Tab. 1. Retigabin (RGB) bei Erwachsenen mit fokalen Epilepsien in den bisherigen klinischen Studien
|
Studiendesign |
Phase |
Anzahl Patienten [N] |
Dosierung |
|
Multizentrische, randomisierte, Plazebo-kontrollierte Doppelblindstudie mit Erwachsenen mit ≥4 fokalen Anfällen pro Monat trotz antikonvulsiver Therapie mit maximal 2 konkurrierenden Antiepileptika (Porter et al., 2007 [32]) |
II |
537 gescreent 399 randomisiert |
Plazebo oder RGB (600, 900 oder 1200 mg) (8 Wochen forcierte Titration, anschließend 8 Wochen Erhaltungsphase ohne Dosisveränderung) |
|
Multizentrische, randomisierte, Plazebo-kontrollierte Doppelblindstudie mit Erwachsenen mit ≥4 fokalen Anfällen pro Monat trotz antikonvulsiver Therapie mit maximal 3 konkurrierenden Antiepileptika mit oder ohne Vagusnervstimulation (VNS) (French et al., 2011 [13]) (RESTORE 1) |
III |
442 gescreent 306 randomisiert |
RGB oder Plazebo wurden titriert bis 1200 mg/d (6 Wochen Titrationsphase); erlaubt war eine Dosisreduktion auf 1050 mg/d am Anfang der 12-wöchigen-Erhaltungsphase |
|
Multizentrische, randomisierte, Plazebo-kontrollierte Doppelblindstudie mit Erwachsenen mit ≥4 fokalen Anfällen pro Monat trotz antikonvulsiver Therapie mit maximal 3 konkurrierenden Antiepileptika (Brodie et al., 2010 [6]) (RESTORE 2) |
III |
696 gescreent 539 randomisiert |
RGB 600 mg/d oder 900 mg/d (4 Wochen Titrationsphase, |
In der Dosisfindungsstudie der Phase II [32] wurden 399 Patienten randomisiert, von denen 96 Plazebo erhielten, 100 Patienten Retigabin 600 mg/Tag, 95 Patienten Retigabin 900 mg/Tag und 106 Patienten Retigabin 1200 mg/Tag. Nach einer 8-wöchigen Beobachtungsphase folgte eine 8-wöchige forcierte Titrationsphase bis zur maximalen Tagesdosis von 600, 900 oder 1200 mg/Tag und anschließend eine 8-wöchige Erhaltungsphase, in der die Dosis nicht verändert werden durfte. Maximal zwei konkurrierende AED waren erlaubt, deren Dosis im Verlauf nicht geändert werden durfte. Ungefähr 70% der Patienten konnten diese 16-wöchige Studienphase beenden. Retigabin zeigte dabei eine dosisabhängige mediane Reduktion monatlicher Anfallsfrequenzen (Plazebo –13,1%, 600 mg/Tag RGB –23,4%, 900 mg/Tag RGB –29,3% und 1200 mg/Tag RGB –35,2%).
Die beiden Zulassungsstudien der Phase III, RESTORE 1 und RESTORE 2, bauten auf dieser Studie auf. RESTORE 1 [13] wurde in den USA, Kanada, Argentinien, Brasilien und Mexiko durchgeführt, während die Daten in RESTORE 2 [6] von therapierefraktären fokalen Epilepsiepatienten aus den USA, Europa, Südafrika und Australien stammen. In beiden Phase-III-Studien wurde ein etwas abweichendes Studiendesign gegenüber der Phase-II-Studie gewählt. Einer 8-wöchigen Beobachtungsphase folgte eine 6-wöchige Titrationsphase in RESTORE 1 (4-wöchig in RESTORE 2) und eine anschließende 12-wöchige Erhaltungsphase. In RESTORE 2 wurden Dosierungen von 600 bzw. 900 mg/Tag Retigabin gewählt, während in RESTORE 1 nur die höchste Zieldosis von 1200 mg/Tag angestrebt wurde. Das Erreichen der Maximaldosis führte bei einigen Patienten dazu, dass die Grenze der Tolerabilität erreicht wurde. Gemäß Studienprotokoll war in der Erhaltungsphase von RESTORE 1 nur eine Dosisreduktion bis 1050 mg/Tag möglich. Innerhalb der sechswöchigen Titrationsphase brachen in RESTORE 1 aufgrund von unerwünschten Ereignissen (AE) ~26% der Patienten, die Verum erhielten, die Studie ab, im Vergleich zu ~8% unter Plazebo. In RESTORE 2 brachen ebenfalls ~26% der Patienten unter 900 mg/Tag Retigabin ab, bei wiederum ~8% unter Plazebo.
Bei den behandelten Patienten in RESTORE 1 und 2 zeigte sich klar eine dosisabhängige Reduktion der Anfallsfrequenz sowohl für die mediane Anfallsreduktion, die von der FDA als relevante Zielgröße angesehen wird (Abb. 2a), als auch für die von der europäischen Zulassungsbehörde (EMA) geforderten Responderraten (Anteil der Patienten mit mindestens ≥50%iger Anfallsreduktion) (Abb. 2b). In den Gruppen, die mit 900 und 1200 mg/Tag behandelt wurden, sprach etwa die Hälfte der Patienten auf die Therapie an. 20 bis 30% der Patienten erfuhren eine mindestens 75%ige Anfallsreduktion und bis zu 7,6% (1200 mg/Tag) wurden anfallsfrei (Abb. 3a und 3b).
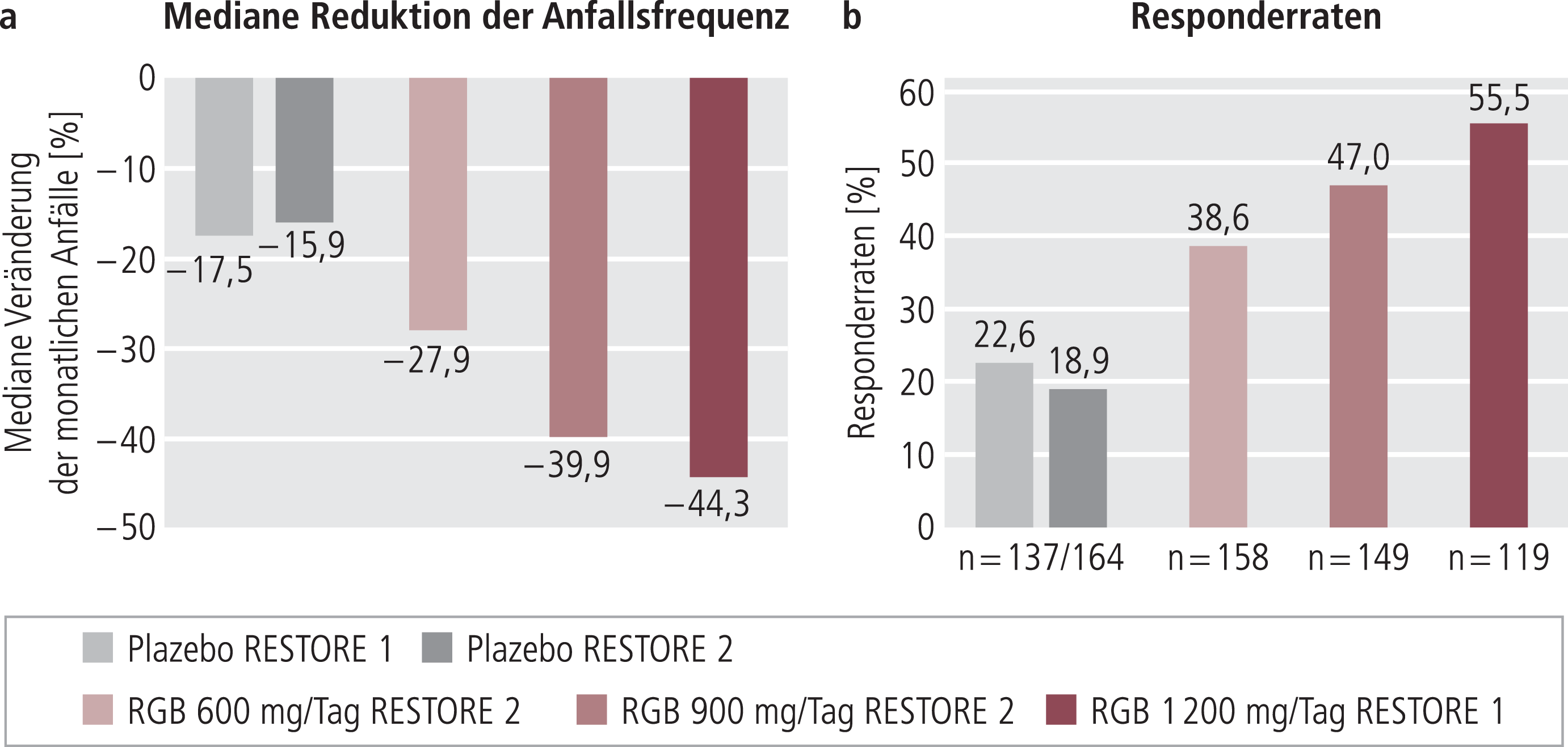
Abb. 2. Wirksamkeit von Retigabin (RGB) in den Phase-III-Studien RESTORE 1 und 2; a) mediane Anfallsreduktion in der Doppelblindphase (FDA-Kriterium); b) mindestens 50%ige Reduktion der Anfallsfrequenz im Vergleich Baseline (Beobachtungsphase vor Medikationsbeginn) vs. Erhaltungsphase (EMA-Kriterium) [6, 13].
Intention-to-treat-(ITT-)Analyse; ITT-Population (FDA): alle randomisierten Patienten, die ≥1-mal Studienmedikation erhielten (= safety population); ITT-Population (EMA): alle randomisierten Patienten, die ≥1-mal Studienmedikation in der Erhaltungsphase erhielten und ≥1 Anfallserhebung aufzeichneten
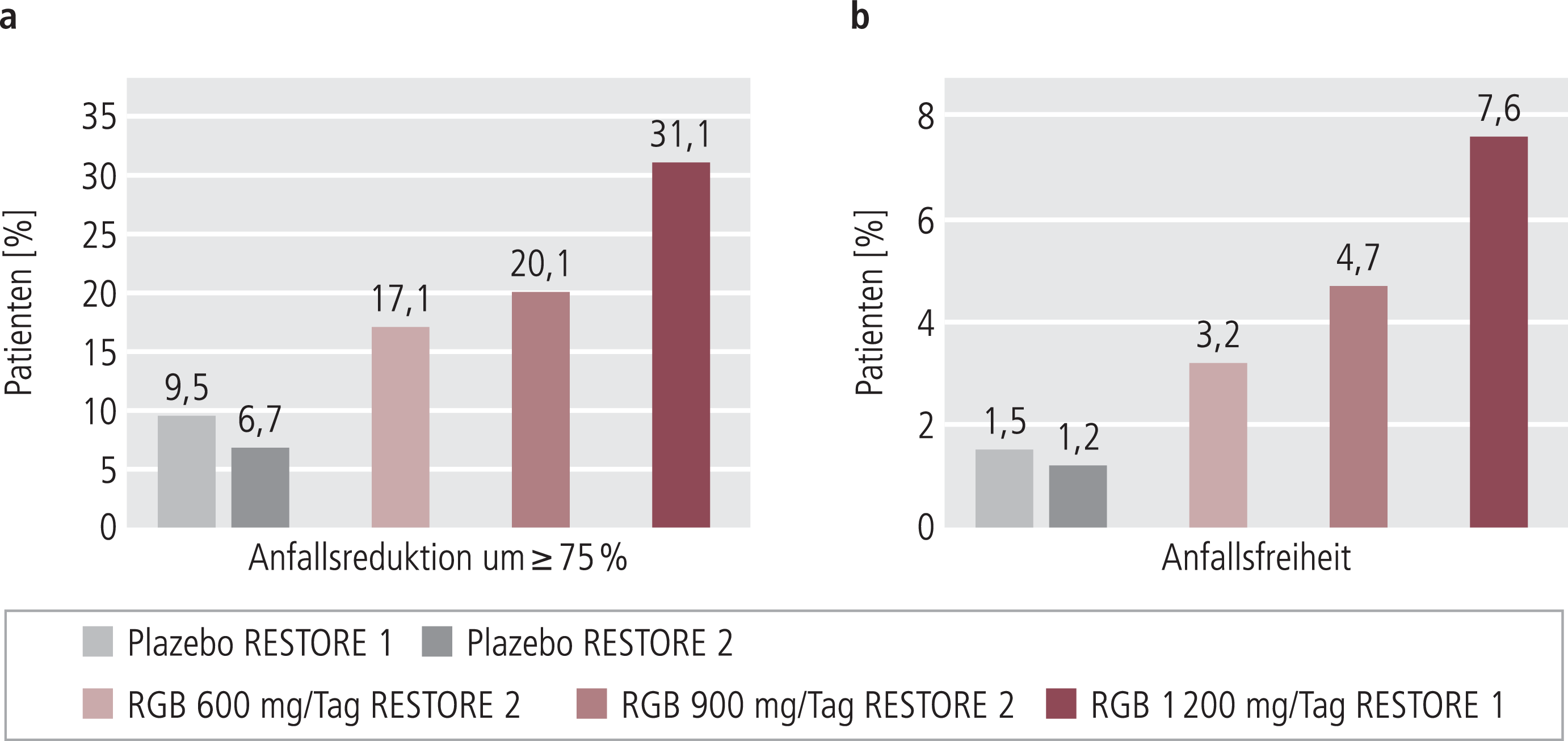
Abb. 3. 75%-Responderraten und Anfallsfreiheit in den beiden Phase-III-Studien RESTORE 1 und 2 [6, 13]; Intention-to-treat-Analyse; Definition ITT-Population (EMA) siehe Abb. 2
Nebenwirkungen
Die meisten Nebenwirkungen unter Retigabin in den oben genannten klinischen Studien [6, 13, 32] waren dosisabhängig und betrafen überwiegend das ZNS. Sehr häufige unerwünschte Wirkungen von Retigabin waren Schwindel, Müdigkeit und Erschöpfung (Tab. 2). Die Nebenwirkungen, die am häufigsten zu einem Absetzen der Behandlung führten, waren Schwindel, Müdigkeit, Erschöpfung und Verwirrtheitszustände. Im Allgemeinen wurden Nebenwirkungen von Retigabin von milder und moderater Intensität beschrieben [6, 13, 32]. Dabei traten die unerwünschten Nebenwirkungen in der Regel innerhalb der ersten acht Behandlungswochen insbesondere während der forcierten Titrationsphase auf. Weder signifikante EKG-Veränderungen noch klinische oder laborchemische Veränderungen, bis auf vereinzelt transient erhöhte Leberfunktionswerte, wurden in RESTORE 1 und 2 berichtet. Allerdings verursachte Retigabin bei einer schnelleren als in der Fachinformation empfohlenen Aufdosierung auf 1200 mg/Tag bei gesunden Probanden eine QT-Verlängerung (durchschnittliche Erhöhung des korrigierten QT-Intervalls von bis zu 6,7 ms) innerhalb von 3 Stunden nach der Einnahme [Fachinformation Trobalt®]. Der Hersteller rät deshalb bei Kombination von Retigabin mit QT-verlängernden Arzneimitteln sowie bei Patienten mit bekanntermaßen verlängertem QT-Intervall, dekompensierter Herzinsuffizienz, ventrikulärer Hypertrophie, Hypokaliämie oder Hypomagnesiämie und bei Patienten ab 65 Jahren zur Vorsicht und zur Ableitung eines EKGs vor Behandlungsbeginn. In den Restharnmessungen wurden leicht erhöhte Restharnmengen bei Patienten mit Retigabin erfasst [13]. Drei Patienten mussten die RESTORE-2-Studie vorzeitig wegen urologischer Nebenwirkungen abbrechen (1×Nephritis, 2×akuter Harnverhalt). Der Hersteller rät deshalb zur Anwendung mit Vorsicht und Aufklärung bei Patienten mit erhöhtem Risiko für einen Harnverhalt. Ebenso gibt es Hinweise auf das Auftreten psychischer Veränderungen unter Retigabin wie Verwirrtheitszustände, psychotische Störungen und Halluzinationen. Eine leichte Gewichtszunahme wurde in den bisherigen Studien beobachtet. Die aktuell verfügbaren Daten schließen die Möglichkeit eines erhöhten Risikos für suizidale Gedanken unter Retigabin nicht sicher aus, was unspezifisch für alle AED zutrifft. Es gilt deshalb besondere Vorsicht bei suizidgefährdeten Patienten. Depressionen traten in den Studien eher seltener unter Retigabin als unter Plazebo (Plazebo: bis 5%, RGB: bis 2%) auf.
Tab. 2. Unerwünschte Nebenwirkungen von Retigabin aus RESTORE 1 und 2
|
Plazebo [%] (n=152/179, RESTORE 1/2) |
RGB 600 mg [%] (n=181, RESTORE 2) |
RGB 900 mg [%] (n=178, RESTORE 2) |
RGB 1200 mg [%] (n=153, RESTORE 1) |
|
|
Schwindel, Benommenheit |
13,8/6,7 |
17,1 |
26,4 |
40,5 |
|
Abnorme Schläfrigkeit |
17,8/10,1 |
14,4 |
26,4 |
31,4 |
|
Müdigkeit |
7,9/2,8 |
17,1 |
15,2 |
15,7 |
|
Verwirrtheit |
2,0/0 |
1,7 |
5,1 |
14,4 |
|
Dysarthrie |
2,0/0 |
5,0 |
1,7 |
12,4 |
|
Kopfschmerzen |
18,4/14,5 |
11,0 |
17,4 |
12,4 |
|
Harnwegsinfektion |
8,6/– |
– |
– |
11,8 |
|
Ataxie |
3,9/– |
– |
– |
11,8 |
|
Verschwommensehen |
2,6/1,7 |
0,6 |
5,1 |
11,8 |
|
Tremor |
3,9/2,2 |
1,7 |
9,0 |
11,1 |
|
Übelkeit |
6,6/2,8 |
8,3 |
7,3 |
10,5 |
|
Sprachstörungen |
0/– |
– |
– |
8,5 |
|
Grippe |
5,3/– |
– |
– |
7,8 |
|
Gedächtnisstörung |
4,6/1,7 |
3,9 |
6,2 |
7,8 |
|
Doppelbilder |
2,6/1,1 |
6,6 |
5,6 |
6,5 |
|
Gangstörung |
2,0/0,6 |
3,3 |
5,1 |
6,5 |
|
Obstipation |
2,0/– |
– |
– |
5,9 |
|
Gleichgewichts- und Koordinationsstörungen |
0,7/1,7 |
6,1 |
5,1 |
5,9 |
|
Blasenentleerungsstörung |
0,7/– |
– |
– |
5,9 |
|
Erbrechen |
5,3/– |
– |
– |
5,2 |
|
Ängstlichkeit |
2,6/– |
– |
– |
5,2 |
|
Dysurie |
1,3/– |
– |
– |
5,2 |
|
Desorientiertheit |
0,7/2,2 |
7,2 |
5,6 |
5,2 |
|
Halluzinationen |
0/– |
– |
– |
4,5 |
Die hier angegebenen Häufigkeiten von Nebenwirkungen beziehen sich auf einzelne Dosierungen in einzelnen Studien und liegen für 1200 mg/Tag manchmal >10% (z.B. für Harnwegsinfekt), obwohl diese in der Fachinformation Trobalt® nicht als sehr häufige Nebenwirkungen angegeben werden (wie auch in dem o. g. Arzneistoffsteckbrief und im Text). Diese scheinbare Diskrepanz kommt dadurch zustande, dass für die Fachinformation gepoolte Daten aus allen drei Phase-II- und -III-Studien herangezogen und gemittelt wurden. Diese gepoolten Daten werden von der Firma GSK noch gesondert veröffentlicht und standen deshalb für diesen Artikel noch nicht zur Verfügung.
Wirkung von Retigabin bei anderen Erkrankungen
Neuronale KV7-Kanäle sind neben der Behandlung von Epilepsien auch potenzielle pharmakologische Targets zur Behandlung anderer Erkrankungen, die mit einer neuronalen Übererregbarkeit einhergehen, wie neuropathische Schmerzen oder Migräne [39]. Experimentelle Daten bei Ratten zeigen, dass Retigabin auch effektiv gegen neuropathische Schmerzen [3], Schmerzchronifizierung bei temporomandibulären Gelenkschmerzen [45] und Dystonie oder paroxysmale Dyskinesien [33] sein könnte. Außerdem suggeriert eine Reihe weiterer Arbeiten einen möglichen vorteilhaften Effekt von Retigabin in der Behandlung von Suchterkrankungen [16], Demenzerkrankungen [14] und Angststörungen [23].
Zusammenfassung
Etwa 30% der Epilepsiepatienten sind pharmakoresistent. Pharmakoresistenz liegt nach der neuen Definition der Internationalen Liga gegen Epilepsie (ILAE) dann vor, wenn trotz zweier „angemessen“ und „adäquat“ eingesetzter AED in Mono- oder Kombinationstherapie weiterhin Anfälle bestehen [24] (Abb. 4). Die Wirkungsmechanismen der bisher auf dem Markt erhältlichen AED sind nicht komplett verstanden. Nach dem aktuellen Wissensstand haben die meisten mehr als einen Wirkungsmechanismus. Eine Reihe wichtiger Wirkungsmechanismen konnte bis dato identifiziert werden, wie die Modulation spannungsabhängiger Ionenkanäle, vor allem die Blockade von Natrium- oder Calciumkanälen, die Erhöhung der inhibitorischen GABA-vermittelten Neurotransmission oder die Dämpfung der exzitatorischen Glutamatwirkung [31].
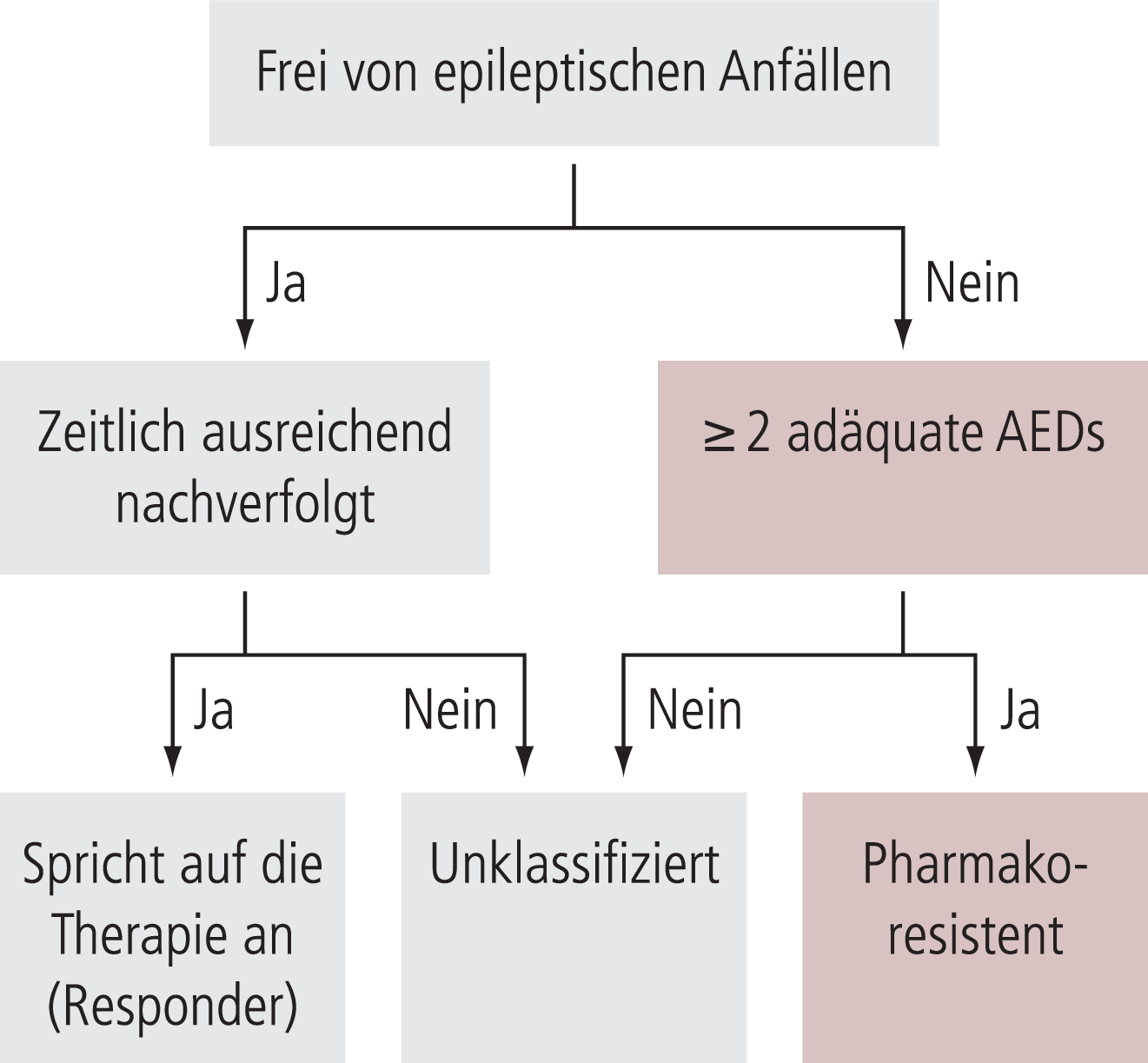
Abb. 4. Klassifikation der therapierefraktären Epilepsiepatienten [mod. nach 24]
Retigabin (USA: Ezogabin) – ein neues AED der dritten Generation – weist einen neuen Wirkungsmechanismus über Kaliumkanäle auf. Es führt zu einer Stabilisierung des offenen Zustands der neuronalen spannungsgesteuerten KCNQ/KV7-Kanäle (KV7.2–KV7.5) durch Bindung im Bereich der Kanalpore [25, 37, 44]. Kardiale KV7.1-Kanäle werden nicht beeinflusst [22, 35, 37, 42, 43]. KCNQ2- und KCNQ3-Mutationen, die zu einem Funktionsverlust KV7.2- und KV7.3-generierter Ströme führen, verursachen eine familiäre Form gutartiger Anfälle bei Neugeborenen [22, 27]. Retigabin führt zu einem Funktionsgewinn über eine Verschiebung der Offenwahrscheinlichkeit von KV7-Kanälen hin zu mehr hyperpolarisierten Membranpotenzialen [26, 35, 42]. Es bedingt also das Gegenteil der Epilepsie-auslösenden Mutationen und vermindert über die Kaliumkanalaktivierung die Feuerungsrate von Nervenzellen.
Retigabin wird schnell resorbiert und erreicht nach etwa 1,5 Stunden die Plasmaspitzenkonzentration. Die terminale Halbwertszeit beträgt 6,7 bis 8,5 Stunden. Für den therapeutischen Dosisbereich von 200 bis 400 mg dreimal täglich besteht eine lineare Dosis-Wirkungs-Beziehung. Retigabin wird durch N-Glucuronidierung und in geringerem Maße durch N-Acetylierung metabolisiert, nicht jedoch durch CYP450-Enzyme. Retigabin und seine Metaboliten werden renal eliminiert. In den Zulassungsstudien fanden sich keine klinisch relevanten pharmakokinetischen Interaktionen von Retigabin und begleitend gegebenen AED (Lamotrigin, Carbamazepin, Phenobarbital, Topiramat, Valproinsäure, Levetiracetam). Eine abschließende Beurteilung der klinischen Relevanz einer Interaktion von Retigabin mit Phenytoin, Carbamazepin und Lamotrigin steht jedoch noch aus.
Für Retigabin wurde die Wirksamkeit in einer Reihe von Tiermodellen für Epilepsie nachgewiesen. In klinischen Phase-II- und -III-Studien zeigte sich eine klar dosisabhängige Reduktion der Anfallsfrequenz sowohl für die FDA-relevante mediane Anfallsreduktion als auch für die von der EMA geforderten Responderraten (Abb. 2a und 2b). Dabei erfuhren 20 bis 30% mindestens eine 75%ige Anfallsreduktion und bis zu 7,6% wurden bei der Maximaldosis von 1200 mg/Tag anfallsfrei (Abb. 3a und 3b). Diese Studien belegen die Wirksamkeit des Kaliumkanalöffners Retigabin in der Behandlung von therapierefraktären Epilepsien. Retigabin könnte zudem bei neuropathischen Schmerzen, Migräne sowie Angst- und Suchterkrankungen wirksam sein.
Retigabin zeigt ein dosisabhängiges Nebenwirkungsprofil. Schwindel, abnorme Schläfrigkeit/Müdigkeit und Erschöpfung treten sehr häufig auf. Unerwünschte Wirkungen im Bereich des Urogenitalsystems (häufig: vermehrtes Auftreten von Harnwegsinfekten, Dysurie, Blasenentleerungsstörungen, Hämaturie, Chromaturie; gelegentlich: Harnverhalt) sind zu beachten. Psychiatrische Nebenwirkungen wie psychotische Störungen/Halluzinationen sowie Sprach- und Sprechstörungen (Dysarthrie, Aphasie, Wortfindungs- und Verständnisstörungen) sind häufig berichtete Nebenwirkungen.
Praktische Hinweise
Es empfiehlt sich, Retigabin entsprechend dem individuellen Ansprechen des Patienten nach Verhältnis von Wirksamkeit und Verträglichkeit aufzudosieren. Dabei sollte nach den bisherigen Erfahrungen bei Therapiebeginn die maximale Gesamttagesdosis bei 3-mal 100 mg/Tag liegen [Fachinformation Trobalt®]. Anschließend wird die Gesamttagesdosis abhängig vom individuellen Ansprechen des Patienten und von der Verträglichkeit um maximal 150 mg pro Woche erhöht. Nach aktueller Datenlage wird die wirksame Erhaltungsdosis zwischen 600 und 1200 mg/Tag erwartet. Die maximal zugelassene Erhaltungsdosis beträgt 1200 mg/Tag, die Unbedenklichkeit und Wirksamkeit von höheren Dosierungen ist nicht nachgewiesen. Bei leicht eingeschränkter Nieren- und Leberfunktion (Creatinin-Clearance 50 bis 80 ml/min; Leberfunktion: Child-Pugh-Score 5 bis 6) wird eine Dosisanpassung für nicht erforderlich angesehen. Bei mittelschwer bis schwer eingeschränkter Nieren- und Leberfunktion (Creatinin-Clearance <50 ml/min; Leberfunktion: Child-Pugh-Score ≥7) werden eine Reduktion der Anfangs- und Erhaltungsdosis von Retigabin um 50% und eine maximale Gesamtdosis von 600 mg/Tag empfohlen [Fachinformation Trobalt®].
Über die Sicherheit und Wirksamkeit von Retigabin bei Kindern und Jugendlichen im Alter von unter 18 Jahren gibt es bisher keine Daten. Ebenso liegen bei Patienten >65 Jahre nur begrenzt Daten vor, weshalb hier eine Dosisreduktion der Anfangs- und Erhaltungsdosis mit maximaler Tagesdosis von 900 mg/Tag empfohlen wird. Ebenso gibt es keine ausreichenden Daten zur Anwendung von Retigabin bei Schwangeren. Retigabin soll nicht während der Schwangerschaft und nicht bei Frauen im gebärfähigen Alter ohne sichere Verhütung angewendet werden.
Danksagung
Die Arbeit wurde von der Robert-Bosch-Stiftung, Stuttgart, und durch das IZEPHA-Projekt 18-0-0, Universität Tübingen, unterstützt.
Literatur
1. Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by 4-aminopyridine in rat entorhinal cortex hippocampal slices. Naunyn Schmiedebergs Arch Pharmacol 1999;359:33–9.
2. Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by low magnesium in rat entorhinal cortex hippocampal slices. Epilepsia 2000;41:28–33.
3. Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol 2003;460:109–16.
4. Borlak J, Gasparic A, Locher M, Schupke H, et al. N-glucuronidation of the antiepileptic drug retigabine: results from studies with human volunteers, heterologously expressed human UGTs, human liver, kidney, and liver microsomal membranes of Crigler-Najjar type II. Metabolism 2006;55:711–21.
5. Brodie MJ, French JA. Management of epilepsy in adolescents and adults. Lancet 2000;356:323–9.
6. Brodie MJ, Lerche H, Gil-Nagel A, Elger C, et al.; RESTORE 2 Study Group. Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology 2010;75:1817–24.
7. Delmas P, Brown DA. Pathways modulating neural kcnq/m (KV7) potassium channels. Nat Rev Neurosci 2005;6:850–62.
8. Dost R, Rundfeldt C. The anticonvulsant retigabine potently suppresses epileptiform discharges in the low Ca2+ and low Mg2+ model in the hippocampal slice preparation. Epilepsy Res 2000;38:53–66.
9. Ferron GM, Patat A, Parks V, Rolan P, et al. Lack of pharmacokinetic interaction between retigabine and phenobarbitone at steady-state in healthy subjects. Br J Clin Pharmacol 2003;56:39–45.
10. Ferron GM, Paul J, Fruncillo R, Richards L, et al. Multiple-dose, linear, dose-proportional pharmacokinetics of retigabine in healthy volunteers. J Clin Pharmacol 2002;42:175–82.
11. Ferron GM, Sachdeo R, Partiot A, Fritz T, et al. Pharmacokinetic interaction between valproic acid, topiramate, phenytoin or carbamazepine and retigabine in epileptic patients. Clin Pharmacol Ther 2001;69:P18.
12. French J, Porter A, Nohria V. Retigabine. In: Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, et al. Progress report on new antiepileptic drugs: a summary of the Seventh Eilat Conference (EILAT VII). Epilepsy Res 2004;61:10–2.
13. French JA, Abou-Khalil BW, Leroy RF, Yacubian EM, et al.; on behalf of the RESTORE 1/Study 301 Investigators. Randomized, double-blind, placebo-controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology 2011;18:1555–63.
14. Gongadze N, Antelava N, Kezeli T, Okudjava M, et al. The mechanisms of neurodegenerative processes and current pharmacotherapy of Alzheimer‘s disease. Georgian Med News 2008;155:44–8.
15. Hammond JMJ. Retigabine. In: Bialer M, Johannessen SI, Levy RH, Perucca E, et al. Progress report on new antiepileptic drugs: a summary of the Tenth Eilat Conference (EILAT X). Epilepsy Res 2010;92:113–6.
16. Hansen HH, Andreasen JT, Weikop P, Mirza N, et al. The neuronal KCNQ channel opener retigabine inhibits locomotor activity and reduces forebrain excitatory responses to the psychostimulants cocaine, methylphenidate and phencyclidine. Eur J Pharmacol 2007;570:77–88.
17. Hempel R, Schupke H, McNeilly PJ, Heinecke K, et al. Metabolism of retigabine (D-23129), a novel anticonvulsant. Drug Metab Dispos 1999;27:613–22.
18. Hermann R, Borlak J, Munzel U, Niebch G, et al. The role of Gilbert‘s syndrome and frequent NAT2 slow acetylation polymorphisms in the pharmacokinetics of retigabine. Pharmacogenomics J 2006;6:211–9.
19. Hermann R, Ferron GM, Erb K, Knebel N, et al. Effects of age and sex on the disposition of retigabine. Clin Pharmacol Ther 2003;73:61–70.
20. Hermann R, Knebel NG, Niebch G, Richards L, et al. Pharmacokinetic interaction between retigabine and lamotrigine in healthy subjects. Eur J Clin Pharmacol 2003;58:795–802.
21. Hiller A, Nguyen N, Strassburg CP, Li Q, et al. Retigabine N-glucuronidation and its potential role in enterohepatic circulation. Drug Metab Dispos 1999;27:605–12.
22. Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 2000;1:21–30.
23. Korsgaard MP, Hartz BP, Brown WD, Ahring PK, et al. Anxiolytic effects of Maxipost (BMS-204352) and retigabine via activation of neuronal Kv7 channels. J Pharmacol Exp Ther 2005;314:282–92.
24. Kwan P, Brodie MJ. Definition of refractory epilepsy: defining the indefinable? Lancet Neurol 2010;9:27–9.
25. Lange W, Geissendörfer J, Schenzer A, Grötzinger J, et al. Refinement of the binding site and mode of action of the anticonvulsant retigabine on KCNQ K+ channels. Mol Pharmacol 2009;75:272–80.
26. Main MJ, Cryan JE, Dupere JR, Cox B, et al. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol 2000;58:253–62.
27. Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch 2010;460:277–88.
28. Mansbach H. Retigabine. In: Bialer M, Johannessen SI, Levy RH, Perucca E, et al. Progress report on new antiepileptic drugs: a summary of the Ninth Eilat Conference (EILAT IX). Epilepsy Res 2009;83:27–31.
29. McNeilly PJ, Torchin CD, Anderson LW, Kapetanovic IM, et al. In vitro glucuronidation of D-23129, a new anticonvulsant, by human liver microsomes and liver slices. Xenobiotica 1997;27:431–41.
30. Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol 2002;61:921–7.
31. Perucca E. An introduction to antiepileptic drugs. Epilepsia 2005;46:31–7.
32. Porter RJ, Partiot A, Sachdeo R, Nohria V, et al.; 205 Study Group. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology 2007;68:1197–204.
33. Richter A, Sander SE, Rundfeldt C. Antidystonic effects of Kv7 (KCNQ) channel openers in the dt sz mutant, an animal model of primary paroxysmal dystonia. Br J Pharmacol 2006;149:747–53.
34. Rostock A, Tober C, Rundfeldt C, et al. D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res 1996;23:211–23.
35. Rundfeldt C, Netzer R. The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells transfected with human KCNQ2/3 subunits. Neurosci Lett 2000;282:73–6.
36. Rundfeldt C. Characterization of the K+ channel opening effect of the anticonvulsant retigabine in pc12 cells. Epilepsy Res 1999;35:99–107.
37. Schenzer A, Friedrich T, Pusch M, Saftig P, et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci 2005;25:5051–60.
38. Straub H, Köhling R, Höhling J, Rundfeldt C, et al. Effects of retigabine on rhythmic synchronous activity of human neocortical slices. Epilepsy Res 2001;44:155–65.
39. Surti TS, Jan LY. A potassium channel, the M-channel, as a therapeutic target. Curr Opin Investig Drugs 2005;6:704–11.
40. Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci 2001;21:5535–45.
41. Tober C, Rostock A, Rundfeldt C, Bartsch R. D-23129: a potent anticonvulsant in the amygdala kindling model of complex partial seizures. Eur J Pharmacol 1996;303:163–9.
42. Wickenden AD, Yu W, Zou A, Jegla T, et al. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol 2000;58:591–600.
43. Wickenden AD, Zou A, Wagoner PK, Jegla T. Characterization of KCNQ5/Q3 potassium channels expressed in mammalian cells. Br J Pharmacol 2001;132:381–4.
44. Wuttke TV, Seebohm G, Bail S, Maljevic S, et al. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol 2005;67:1009–17.
45. Xu W, Wu Y, Bi Y, Tan L, et al. Activation of voltage-gated KCNQ/Kv7 channels by anticonvulsant retigabine attenuates mechanical allodynia of inflammatory temporomandibular joint in rats. Mol Pain 2010;6:49.
Abkürzungen
AE: adverse event, AED: antiepileptic drug, BFNS: benigne familiäre Neugeborenenanfälle (benign familial neonatal seizures), EGB: ezogabine/Ezogabin, EMA: European Medicines Agency, FDA: Food and Drug Administration, GABA: gamma amino butyric acid (Gamma-Aminobuttersäure), ILAE: Internationale Liga gegen Epilepsie, LQTS1: Long-QT-Syndrom Typ 1, RGB: retigabine/Retigabin
Prof. Dr. med. Holger Lerche, Gökce Orhan, Abteilung Neurologie mit Schwerpunkt Epileptologie, Hertie-Institut für Klinische Hirnforschung, Universität Tübingen, Hoppe-Seyler-Straße 3, 72076 Tübingen, E-Mail: holger.lerche@ uni-tuebingen.de
Dr.med. Thomas V. Wuttke, MGH-HMS Center for Nervous System Repair, Departments of Neurosurgery and Neurology, and Program in Neuroscience, Harvard Medical School, Boston, MA 02114, USA; Nayef Al-Rodhan Laboratories, Massachusetts General Hospital, Boston, MA 02114, USA
Priv.-Doz. Dr.rer.nat. Anne T. Nies, Dr. Margarete Fischer-Bosch-Institut für Klinische Pharmakologie, Stuttgart, Auerbachstraße 12, 70376 Stuttgart, und Universität Tübingen
Prof. Dr. med. Matthias Schwab, Dr. Margarete Fischer-Bosch-Institut für Klinische Pharmakologie, Stuttgart, und Abteilung Klinische Pharmakologie, Institut für Experimentelle und Klinische Pharmakologie und Toxikologie, Universitätsklinikum Tübingen, Otfried-Müller-Straße 45, 72076 Tübingen
The KCNQ/KV7 potassium channel opener Retigabine as add-on therapy for partial epilepsy. Pharmacological and clinical data
This review summarizes the pharmacological, preclinical and clinical data on the new anticonvulsant compound retigabine (RGB) and discusses the impact of this drug for patients with refractory partial epilepsy. Retigabine increases the activity of voltage-gated KCNQ/KV7 channels and thus exhibits a novel mechanism of action as compared to available anticonvulsive drugs. Retigabine has been investigated in three multicenter, randomized, double-blind and placebo-controlled clinical phase II and III trials in adults with ≥4 partial-onset seizures per month and showed efficacy in pharmacoresistant epilepsy. In march 2011, Retigabine received approval in Europe as Trobalt®.
Key words: Epilepsy, potassium channel, pharmacotherapy, antiepileptic drug, retigabine
Psychopharmakotherapie 2011; 18(04)