Hans-Peter Volz, Werneck, Jürgen Staedt, Berlin-Spandau, Ion-George Anghelescu, Berlin-Charlottenburg, und Eckart Rüther, München
Generika spielen als kostengünstige Variante eines Originalpräparats eine wichtige Rolle in unserem Gesundheitssystem. In der Regel werden Originalpräparate, sobald kostengünstigere Generika auf den Markt kommen, in breitem Umfang durch die deutlich kostengünstigeren Generika innerhalb der kassenärztlichen Versorgungspraxis ersetzt.
Wenig bekannt ist den meisten Anwendern, dass Generika und Originalpräparate keineswegs gleich sind, sondern dass zum Teil durchaus Abweichungen des Generikums vom Originalpräparat existieren, die die sichere und effektive Anwendung des Arzneimittels beeinflussen können. Im Folgenden sollen die wichtigsten Punkte hierzu dargestellt werden.
Definitionen
Um die Zulassung für ein Generikum in Deutschland oder in Europa zu erhalten, muss der Hersteller den Nachweis der Bioäquivalenz zum Originalpräparat erbringen [2]. Die Bioäquivalenz setzt sich aus der pharmazeutischen Äquivalenz und der Bioverfügbarkeit zusammen.
Pharmazeutische Äquivalenz bedeutet, dass zwei Medikamente denselben Arzneistoff in derselben molaren Menge (Dosis) und der gleichen Darreichungsform enthalten und gleiche Standards in Bezug auf die Qualität haben, wobei es Unterschiede in bestimmten Charakteristika (z.B.Form, Farbe und Geschmack, Hilfsstoffe) geben kann [1].
Die Bioverfügbarkeit ist eine pharmakologische Messgröße für den Anteil eines Stoffs, der unverändert im systemischen Kreislauf zur Verfügung steht. Das heißt, sie gibt an, wie schnell und in welchem Umfang der Stoff resorbiert wird und am Wirkort zur Verfügung steht. Eine orale Bioverfügbarkeit von 100% bedeutet beispielsweise, dass die gesamte oral absorbierte Menge tatsächlich im Blutkreislauf erscheint.
Gemäß der Note for Guidance der European Medical Agency (EMEA), die die regulatorische Grundlage für die Zulassung von Generika bildet, kommt den folgenden Parametern entscheidende Bedeutung zu (siehe Abb. 1):
- „Area under the Curve“ (AUC), entsprechend der Wirkstoffgesamtmenge, die der Körper aufgenommen hat
- Plasmaspitzenkonzentration (Cmax), entsprechend der maximalen Konzentration, die im Verlauf im Plasma erhoben wird
- Zeit bis zum Erreichen der Plasmaspitzenkonzentration (tmax)
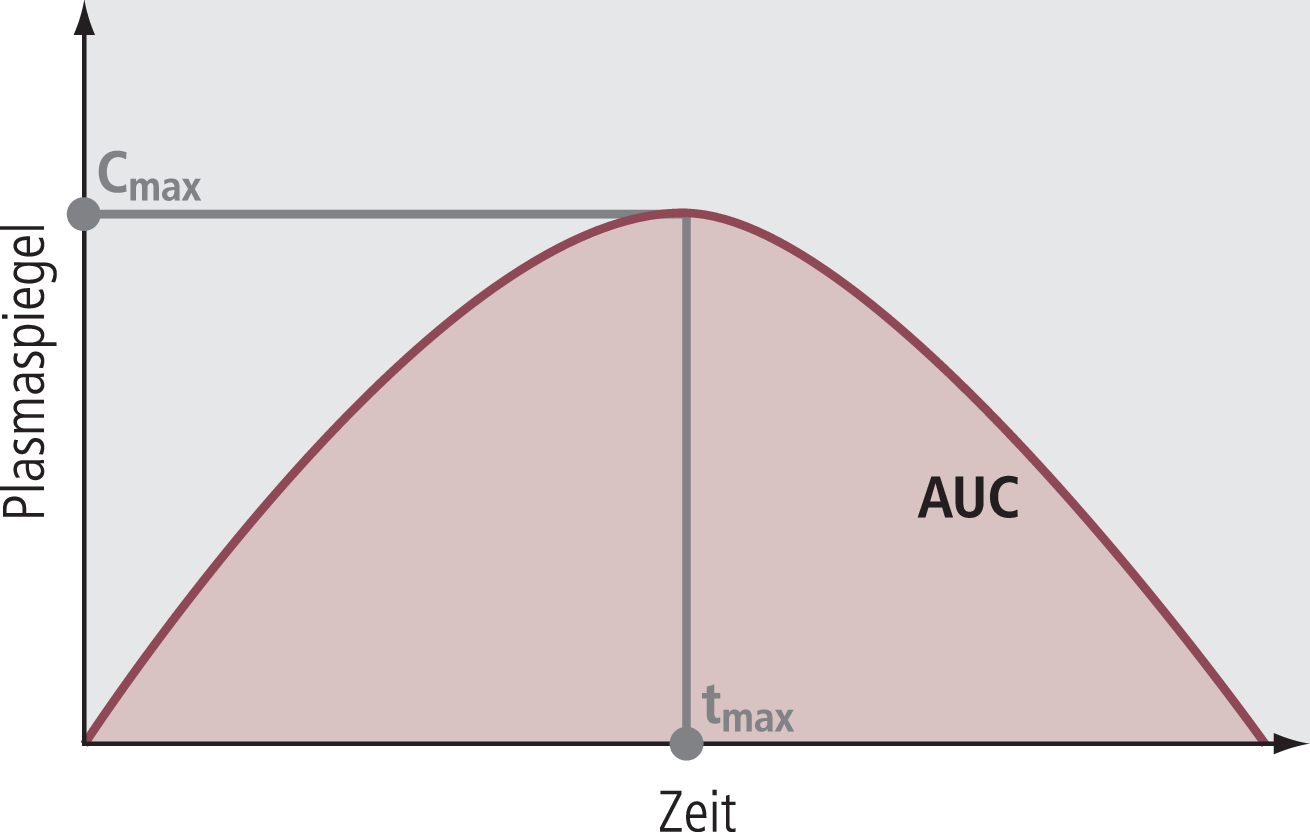
Abb. 1. Darstellung der wichtigsten pharmakokinetischen Parameter Die dunkelrote Linie repräsentiert den Verlauf der Plasmakonzentration über die Zeit; Cmax ist die maximale Plasmakonzentration, tmax ist der Zeitpunkt, zu dem die maximale Plasmakonzentration erreicht wird. Die AUC ist die Fläche unter der Plasmaspiegel-Zeit-Kurve als Maß für die Wirkstoffgesamtmenge, die der Körper aufgenommen hat.
Dieses so definierte Konstrukt (ohne die tmax) dient als Basis der Messung von Bioäquivalenz.
(Die letzten beiden Parameter sind insbesondere bei Retard-Präparaten von Bedeutung, siehe weiter unten.)
Im Gegensatz zur Bioäquivalenz als notwendige Voraussetzung für die Zulassung von Generika bedeutet therapeutische Äquivalenz die Gleichwertigkeit zweier Präparate mit gleichem Wirkstoff und Wirkstoffprinzip in der klinischen Anwendung bezüglich Wirksamkeit und Unbedenklichkeit beziehungsweise Tolerabilität [16]. Die Bioäquivalenz stellt pharmakokinetische Parameter in den Mittelpunkt, die therapeutische Äquivalenz die klassischen klinischen Parameter wie Wirksamkeit, Verträglichkeit und Sicherheit eines Arzneimittels. Die therapeutische Äquivalenz muss bei der Zulassung eines Generikums nicht nachgewiesen werden [2]. Somit werden zur Zulassung eines Generikums keine klinischen Studien, sondern nur pharmakokinetische Studien verlangt (wie diese aufgebaut sein müssen, wird weiter unten dargelegt). Dies ist aus pharmakologischer Sicht auch berechtigt, unter anderem, da die Wirkung eines Wirkstoffs von seinem pharmakokinetischen Verhalten determiniert wird.
Zulassungsvoraussetzungen
In der Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 (Art. 10, Abs. 2b) wird ausgeführt, welchen Anforderungen ein Generikum standhalten muss, um in der EU zugelassen zu werden: Das Generikum muss im Vergleich zum Originalpräparat
- die gleiche qualitative und quantitative Zusammensetzung aus Wirkstoffen,
- die gleiche Darreichungsform sowie
- Bioäquivalenz zum Originalpräparat (demonstriert in einer Bioäquivalenzstudie)
aufweisen [2].
Der Nachweis der Bioäquivalenz gilt dabei als Surrogatparameter für therapeutische Äquivalenz, da die Durchführung großer Studien zum Nachweis der therapeutischen Äquivalenz aufgrund des erheblich höheren Aufwands nur in wenigen Ausnahmefällen stattfindet. Wie bereits dargelegt, sind die Zielparameter einer Bioäquivalenzstudie aber pharmakokinetische, nicht etwa klinische Maße. Aus pharmakologischer Sicht wird – wie bereits erwähnt – die Wirkung einer Substanz von ihrem pharmakokinetischen Verhalten bestimmt.
Bioäquivalenzprüfungen werden in der Regel mit gesunden, meist männlichen Probanden im Alter zwischen 18 und 55 Jahren durchgeführt [2]. Empfohlen werden zudem Normalgewicht sowie Nichtraucher-Status. Vorgeschrieben ist ein Minimum von zwölf Probanden. Die Hauptziele einer solchen Studie sind wie folgt: Das 90%-Konfidenzintervall der relativen mittleren AUC (Area under the Curve) des Generikums in Relation zum Originalpräparat soll zwischen 80 und 125% liegen (zur Illustration dieses statistischen Maßes siehe Abb. 2). Die relative mittlere Cmax des Generikums zum Originalpräparat soll zwischen 80 und 125% betragen.
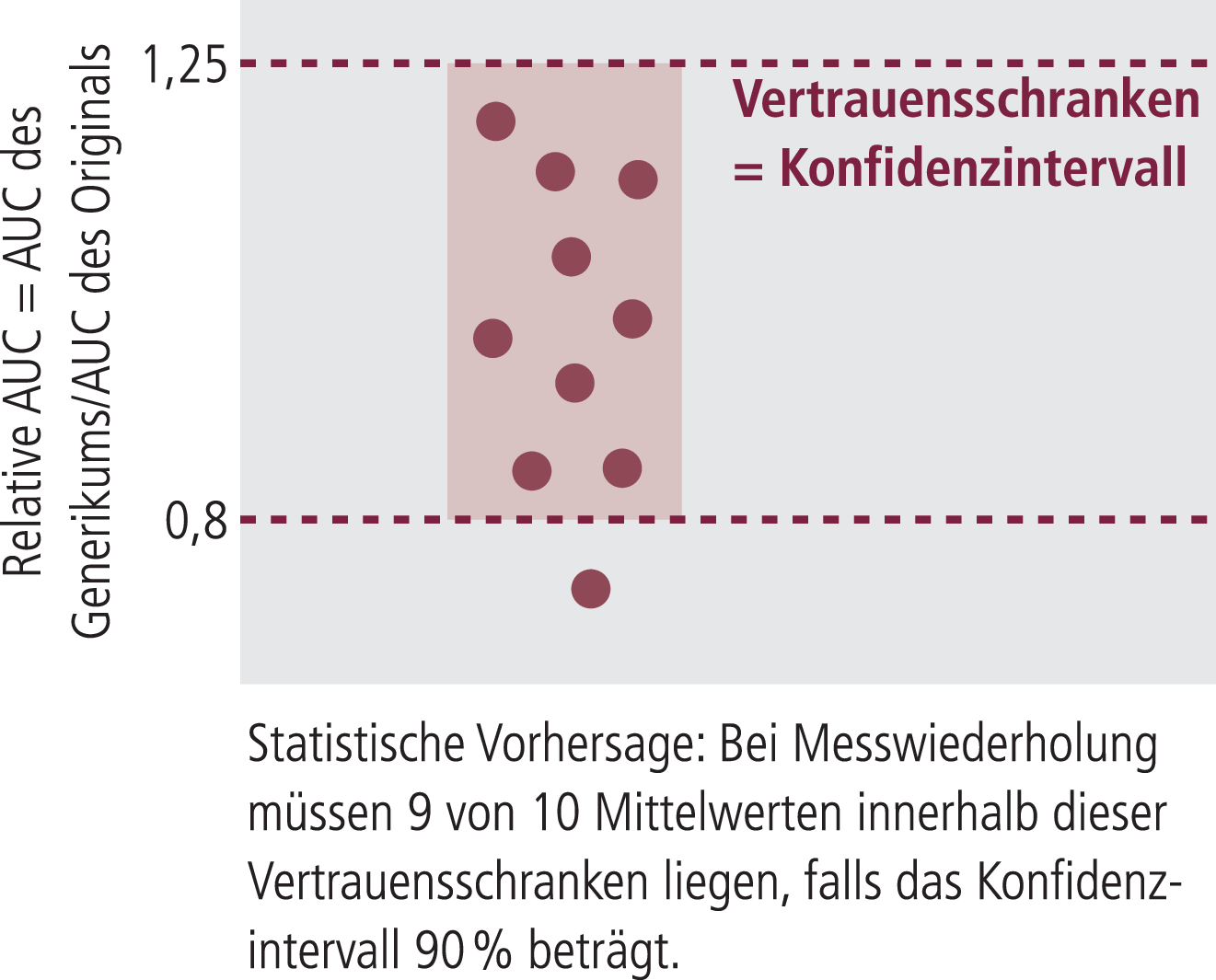
Abb. 2. Bedeutung der relativen AUC, die bei Bioäquivalenzstudien zwischen 0,8 und 1,25 bei einem Konfidenzintervall von 90% schwanken darf
In der Abbildung 3 ist die in Bioäquivalenzstudien zugelassene Schwankungsmöglichkeit für die AUC basierend auf der Darstellung der Abbildung 1 illustriert. Relativierend ist hier anzufügen, dass es auch bei verschiedenen Chargen eines Originalpräparats zu Schwankungen der Bioverfügbarkeit kommt, wobei diese Schwankungen in der Regel aber wesentlich geringer ausgeprägt sind als die hier definierte maximal mögliche Schwankungsbreite.
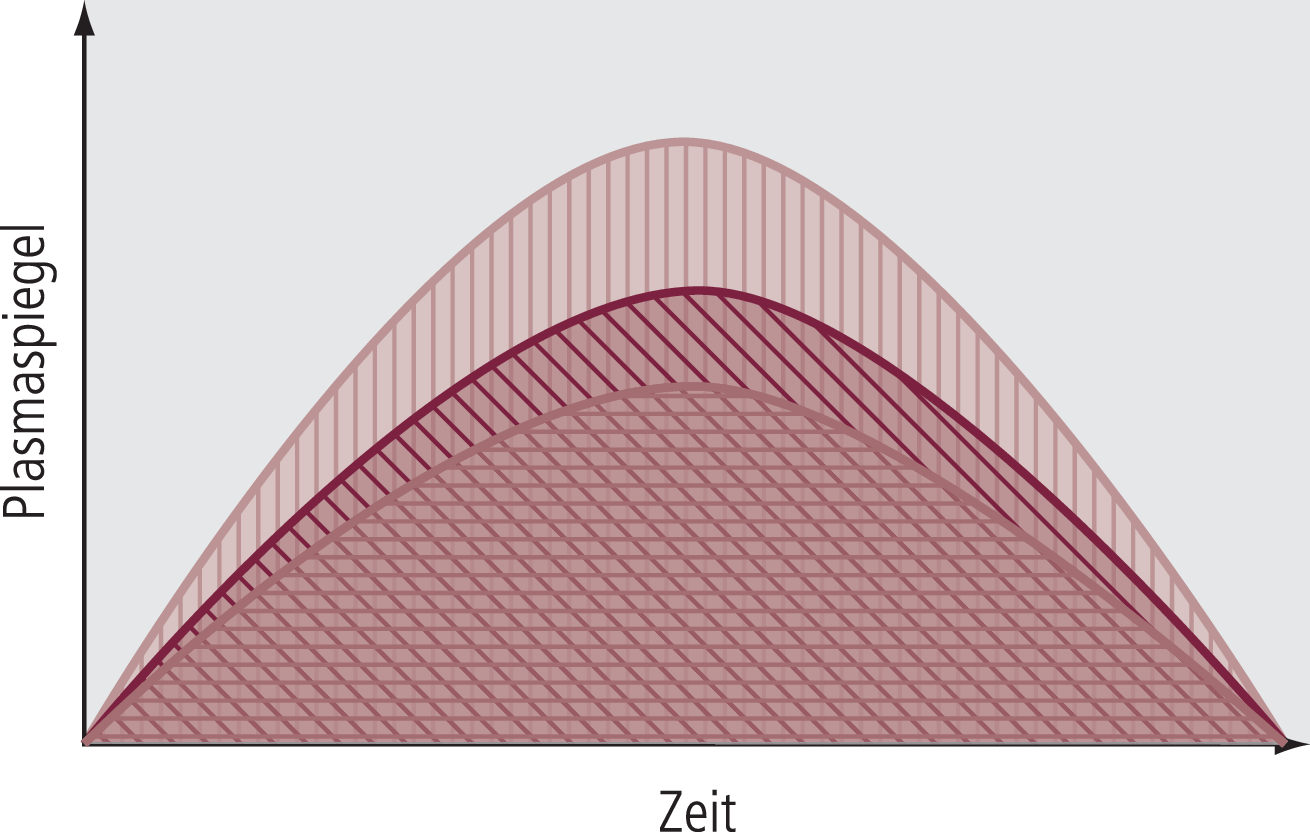
Abb. 3. Darstellung der durch die EMEA-Note for Guidance zugelassenen Schwankungsmöglichkeiten in der AUC zwischen Originalpräparat (die dunkelrote Linie repräsentiert die Plasma-Konzentration über die Zeit, die schräg schraffierte Fläche die AUC) und einem Generikum, das sich in der AUC zwischen der unteren durchgezogenen Linie (AUC 80% als unteres Limit) und der oberen durchgezogenen Linie (AUC 125% als oberes Limit) befinden muss
Problemfelder
Aus diesen Vorschriften resultieren folgende Fragen in Bezug auf den Einsatz eines Generikums anstatt des Originalpräparats:
- Was sind die Auswirkungen dieser gesetzlich erlaubten Schwankungen?
- Wie wirken sich unterschiedliche Hilfsstoffe aus?
- Sind die Ergebnisse der Bioäquivalenzprüfungen statistisch sicher?
- Welche Rolle spielt eine interindividuelle und intraindividuelle Variabilität?
- Sind die Ergebnisse von Studien mit jungen, gesunden Probanden auf alle Patientinnen und Patienten übertragbar?
(Unbenommen ist allerdings auch der Punkt, dass Ergebnisse aus Phase-II- oder -III-Studien auch nur eingeschränkt auf die Routineversorgung übertragen werden können.)
Zu diesen Fragen in Bezug auf die Methodik der Sicherung der „Äquivalenz“ eines Generikums und der Übertragbarkeit der so gewonnen Ergebnisse auf unterschiedliche Patienten ergibt sich auch noch die Frage, welche Auswirkungen Unterschiede in der Verpackung, dem Aussehen, dem Geschmack oder dem Namen auf die Compliance, speziell bei vulnerablen Patientengruppen, haben.
Die Deutsche Pharmazeutische Gesellschaft hat zu diesem Thema eine Leitlinie veröffentlicht [1] und geht folgendermaßen auf die Problematik ein: „Bei der Anwendung von Generika – und analog auch beim Einsatz von Re- und Parallelimporten – sind verschiedene Szenarien von praktischer Relevanz, die mit Blick auf die Sicherstellung des gewünschten therapeutischen Erfolgs unterschiedlich kritisch zu bewerten sind.“ Besondere Vorsicht ist bei kritischen Medikamenten geboten, beispielsweise mit enger therapeutischer Breite, komplexem Therapieregime, bedeutenden Wechselwirkungen oder individueller Dosisanpassung. Arzneimittelgruppen, bei denen eine Substitution als schwierig angesehen wird, sind unter anderem häufig verwendete Psychopharmaka wie Antidepressiva oder Neuroleptika (Tab. 1) [1]. Hierbei handelt es sich um Medikamente, die bei schweren und potenziell lebensbedrohlichen Erkrankungen eingesetzt werden, wobei eine zuverlässige und stabile Einstellung für einen langfristigen Therapieerfolg von entscheidender Bedeutung ist.
Tab. 1. Arzneimittelgruppen, bei denen eine Substitution besonders kritisch sein kann [1]
|
Antiarrhythmika |
|
Antiasthmatika |
|
Antidementiva |
|
Antidepressiva |
|
Antidiabetika |
|
Antiepileptika |
|
Antikoagulanzien |
|
Antiparkinsonmittel |
|
Herzwirksame Glykoside |
|
Hormonale Kontrazeptiva |
|
Immunsuppressiva |
|
Interferone |
|
Neuroleptika |
|
Opioid-Analgetika |
|
Thrombozytenfunktionshemmer |
|
Zytostatika |
Die besondere Nennung von bestimmten Psychopharmaka in diesem Kontext hängt unseres Erachtens nicht mit Faktoren wie einer besonders geringen therapeutischen Breite von Antidepressiva oder Neuroleptika zusammen (diese ist – im Gegenteil – bei modernen Vertretern dieser Substanzgruppen sogar relativ groß), als vielmehr mit der Frage, welcher individuelle Plasmaspiegel für den individuellen Patienten den geeigneten Kompromiss zwischen ausreichender Wirksamkeit und tolerierbaren Nebenwirkungen darstellt.
Vor allem bei Arzneimitteln mit enger therapeutischer Breite und/oder hoher Toxizität – beispielsweise Lithiumsalzen, Immunsuppressiva oder einigen Antiepileptika, die eine individuelle Anpassung bei jedem Patienten benötigen – können schon kleine Unterschiede in der Verfügbarkeit des Wirkstoffs im Körper zu Konsequenzen, wie vermehrten Nebenwirkungen oder einer geringeren Wirkung, führen. Bei Medikamenten mit geringer therapeutischer Breite kann also nicht unbedingt mit identischer Wirksamkeit gerechnet werden.
Die Therapie bei Patienten mit psychischen Erkrankungen erfolgt meist durch eine längerfristige Medikamenteneinnahme. Wechsel zwischen Originalpräparat und Generika oder zwischen verschiedenen Generika können dabei zu deutlichen Plasmaspiegelschwankungen führen. Die Plasmakonzentrationen können bei einem Generikum bis zu 25% höher oder 20% niedriger sein als bei dem entsprechenden Originalpräparat, das als Referenz diente. Beim Vergleich einzelner Generika untereinander können die Abweichungen sogar so groß sein, dass keine Bioäquivalenz mehr vorliegt, beispielsweise wenn eines der Präparate einen Plasmaspiegel von 80% im Vergleich zum Original hat und ein anderes 120%. Bei der chronischen Medikamentengabe können diese Abweichungen in Abhängigkeit von den pharmakologischen Eigenschaften von Bedeutung sein und die Wirksamkeit beeinflussen. Hinzu kommt die intraindividuelle Variabilität, die den Unsicherheitsfaktor zusätzlich erhöht. Da in der täglichen Praxis häufige Medikamentenwechsel durchaus die Regel sind, kann dies negative Auswirkungen auf den Therapieerfolg haben.
Abgesehen von objektiven Gesichtspunkten sollte zudem in bestimmten Situationen auf eine Substitution verzichtet werden, selbst wenn fachlich aus Apothekersicht ein Austausch verantwortbar scheint: Bei ängstlichen oder agitierten Patienten kann die Substitution zur Einnahmeverweigerung oder zu einer gravierenden Verschlechterung der Compliance führen. „Die Substitution könnte beim Patienten Befürchtungen auslösen, dass sich sein Krankheitsbild verschlechtern könnte. Dabei ist es unerheblich, ob die Ängste rational begründet sind oder nicht“ [1].
Galenik
Ein weiterer wichtiger Aspekt eines Arzneimittels ist die galenische Zubereitung wie beispielsweise Retardformulierungen. Unter Galenik wird die Wissenschaft von der Zubereitung von Arzneimitteln aus Arznei- und Hilfsstoffen verstanden. In der bereits erwähnten Leitlinie „Gute Substitutionspraxis“ zählen Retardpräparate zu den Darreichungsformen, bei denen eine Substitution kritisch sein kann. So heißt es in dieser Leitlinie: „Bei Präparationen mit modifizierter Wirkstofffreisetzung wird beispielsweise die Resorption aus dem Magen-Darm-Trakt ganz entscheidend durch die Eigenschaften des Produktes determiniert.“ Der Hauptgrund hierfür ist, dass Retardpräparate durch die verzögerte und verlängerte, möglichst gleichmäßige Wirkstofffreisetzung Fluktuationen der Wirkstoffkonzentration im Plasma vermindern, die „Täler“ (troughs) und „Spitzen“ (peaks) geringer ausgeprägt sind. Dies ist nur mit einer sehr speziellen Galenik möglich. Die Zulassungsvoraussetzungen für Generika fordern zwar eine gleiche Darreichungsform, erlauben jedoch unterschiedliche Techniken, um Retardformulierungen herzustellen. Beispielsweise wird für die Retardformulierung eines in Kanada zugelassenen Generikums von Venlafaxin (Novo-Venlafaxin; Hersteller TEVA) eine sogenannte Single-Unit-Technologie (z. B. überzogene Tablette oder Kapsel) verwendet, während für das Original vom Hersteller eine Mikrosphärentechnologie (Mikrokapseln in einer Kapsel) verwendet wird [11].
Zur Evaluierung der Bioäquivalenz der beiden Formulierungen Novo-Venlafaxin und Trevilor® retard wurde eine verblindete, randomisierte Cross-over-Studie mit 24 männlichen, gesunden Probanden im Alter zwischen 18 und 55 Jahren unter Nüchternbedingungen und nach Nahrungsaufnahme durchgeführt. Nach der Einmaleinnahme von 150 mg unter Nüchternbedingungen ergaben die erhobenen Bioäquivalenzparameter für Novo-Venlafaxin eine Wirkstoffgesamtdosis (AUC) von 113,5% und eine Spitzenplasmakonzentration (Cmax) von 124,5% gegenüber dem Originalpräparat und lagen somit im erlaubten Intervall von 80 bis 125% [11]. Auffallend war, dass unter Novo-Venlafaxin eine 2-fach höhere Rate an gastrointestinalen Nebenwirkungen beobachtet wurde. In-vitro-Daten zeigen eine schnellere Wirkstofffreisetzung von Novo-Venlafaxin mit höheren Spitzenkonzentrationen vor allem in den ersten 2 bis 4 Stunden (Data on file). Diese Beobachtungen im Kontext der unterschiedlichen Bioäquivalenzparameter können die erhöhte Rate an gastrointestinalen Nebenwirkungen von Novo-Venlafaxin erklären.
Der klinische Alltag
Die Frage ist, ob die Befürchtungen einiger Experten für den klinischen Alltag von Relevanz sind. In der Literatur finden sich tatsächlich verschiedene Beispiele dafür, dass der Wechsel von einem Originalpräparat auf ein Generikum nicht immer so reibungslos vonstatten geht, wie man das bei Äquivalenz (=Gleichwertigkeit) erwarten sollte. Es gibt Angaben, dass etwa 10% der Patienten Probleme nach dem Wechsel von einem Originalpräparat auf ein Generikum zeigen [12, 15]. Dies kann sowohl einen Wirksamkeitsverlust als auch verstärkte Nebenwirkungen beinhalten, wobei der sogenannte Nozebo-Effekt (der Patient reagiert subjektiv auf den Wechsel, obwohl objektiv Gleichwertigkeit besteht) nicht immer auszuschließen ist.
Beispiel Epilepsie
Eine Arzneimittelgruppe, bei der ein unkontrollierter Wechsel sehr kritisch gesehen wird, sind die Antiepileptika. In den 90er Jahren kam es international vermehrt zu Fallberichten über zunehmende/neu aufgetretene epileptische Anfälle im Anschluss an einen Präparatewechsel [3, 5, 7, 13, 14, 18].
Daraufhin veröffentlichte 2002 eine Ad-hoc-Kommission der Deutschen Gesellschaft für Epileptologie den Vorschlag, klassische Antiepileptika nicht in die Aut-idem-Regelung aufzunehmen, da Umstellungen von anfallsfreien Patienten generell unterbleiben sollten [9]. Zeitgleich wurden die Antiepileptika von der Deutschen Pharmazeutischen Gesellschaft in der Leitlinie „Gute Substitutionspraxis“ in die Gruppe der für eine Substitution kritischen Medikamente aufgenommen [1]. In der Folge wurden Antiepileptika nicht in die modifizierte Aut-idem-Regulation von 2003 mit einbezogen. Am 1. April 2008 änderte sich jedoch die Situation durch den Rahmenvertrag zwischen den Spitzenverbänden der GKV und dem Deutschen Apothekerverband: Bei fehlendem Aut-idem-Kreuz wurde auch bei Antiepileptika die Substitution vorgeschrieben, wobei die Präparatewahl dem Apotheker überlassen wurde. Dies führte im April 2008 zu einer erneuten Stellungnahme der Deutschen Gesellschaft für Epileptologie: Der unkontrollierte Wechsel sei nicht zu verantworten, und deshalb erfolgte ein Aufruf an die Verordner, „aut-idem“ anzukreuzen [8].
Beispiel Antithrombotika (Warfarin)
Aus Israel liegt eine Sekundärdatenanalyse von 975 Patienten vor, die zunächst das Originalpräparat für sechs Monate einnahmen und dann für sechs Monate auf ein in den Hilfsstoffen modifiziertes Präparat wechselten. Dabei zeigte sich in der zweiten Periode, dass die mittlere Dosis des Medikaments um 26,5% höher und der INR (International Normalized Ratio) um 14,7% niedriger (p<0,001) lagen. Bei 94 Patienten ohne Dosisanpassung fiel der INR auf subtherapeutisches Niveau [4].
Beispiel Schizophrenie (Clozapin)
Aus dem Bereich Schizophrenie gibt es ein Praxisbeispiel von sieben vor Umstellung stabilen Schizophrenie-Patienten einer Pflegeeinrichtung, die ohne Wissen der Patienten, des Arztes oder der Pflegekräfte vom Originalpräparat auf generisches Clozapin umgestellt wurden.
Nach dem Wechsel kam es bei allen sieben Patienten zu einem raschen und schweren Rückfall, der in fünf Fällen sogar eine Hospitalisation notwendig machte. Nach dem Rückwechsel auf das Originalpräparat bildeten sich die Symptome zurück [10]. Auch andere Beobachtungen bestätigen die Beschreibungen dieser Fallserie [6]. Die abschließende Bewertung dieser Fallserie ist aber – da keine Plasmaspiegelbestimmungen vorliegen – nur schwer möglich.
Beispiel Angstzustände (Citalopram)
Aus dem Bereich der Neurologie/Psychiatrie gibt es das Beispiel von 20 Angstpatienten, die stabil auf ein Originalpräparat eingestellt waren. Bei diesen Patienten kam es unerwartet zu einer unerklärlichen Verschlimmerung der Symptome und teilweise auch zur Zunahme der Nebenwirkungen. Folgende Erklärung ergab sich: Der Apotheker hatte die Patienten ohne ihr Wissen und ohne Wissen des Arztes auf ein Generikum umgestellt. Die Symptome begannen vier Tage nach Umstellung, die Erholung nach der Rückumstellung auf das Original hingegen dauerte über drei Wochen [17]. Auch bei dieser Untersuchung lagen keine Plasmaspiegelbestimmungen vor, so dass die Interpretierbarkeit eingeschränkt ist. Zudem handelt es sich, wie auch bei den Clozapin-Untersuchungen (siehe oben) um Untersuchungen im Rahmen von Versorgungsstudien, bei denen keine strukturierten Messinstrumente angewendet, sondern die Wirkungen und Nebenwirkungen im Rahmen der routinemäßigen klinischen Evaluation erhoben wurden.
Fazit
Generika haben ohne Zweifel ihren Platz im deutschen Gesundheitssystem. Ein Präparatewechsel sollte jedoch immer mit Bedacht erfolgen. Dies gilt insbesondere für Substanzen mit
- enger therapeutischer Breite,
- bestimmter Kinetik/Galenik (z. B. bei schwer herzustellenden Retardformulierungen),
- erforderlicher individueller Dosiseinstellung (v. a. bei erschwertem Monitoring) und
- schweren Konsequenzen im Falle des Therapieversagens sowie für Hochrisikogruppen mit leicht gefährdeter Compliance.
Diese Gesichtspunkte sollten bei der individuellen Abschätzung, ob eine Substitution sinnvoll ist, unbedingt berücksichtigt werden. Falls Umstellungen erfolgen, sollte der Patient darüber aufgeklärt werden, dass es sich um wirkstoffgleiche Medikamente handelt, auch wenn die Packung und meist auch die Tablette selbst unterschiedlich aussehen.
Ein weiterer wichtiger Gesichtspunkt ist, ob bei der Einführung von Generika strengere Maßstäbe angelegt werden sollten, um einen Teil der geschilderten Probleme zu vermeiden. So könnten beispielsweise engere Grenzen für die Feststellung der Bioäquivalenz (z. B. <10%) festgelegt werden. Aus klinischer Sicht – wenngleich wahrscheinlich wirklichkeitsfremd – wären klinische Studien, sogenannte „non inferiority-trials“, mit Patienten wünschenswert, um die tatsächliche „therapeutische Äquivalenz“ im Vergleich zum Originalpräparat zu belegen. Zudem könnten prospektiv geplante Datenbank-gestützte systematische Erhebungen beziehungsweise Vergleiche zwischen Generika und Original zur Feststellung vor allem von Verträglichkeitsunterschieden genutzt werden. Insgesamt könnten derartige Maßnahmen dazu beitragen, die Risiken bei einem Wechsel von einem Originalpräparat auf ein Generikum oder bei einem Wechsel innerhalb der Generika zu minimieren und somit den Therapieerfolg sicherzustellen.
Literatur
1. Blume H, Brauer KG, Dingermann T, Mutschler E, et al. Gute Substitutionspraxis (GSP). Dtsch Apoth Ztg 2002;142:1205–14.
2. EMEA. Note for guidance on the investigation of bioavailability and bioequivalence. 2001.
3. Gilman JT, Alvarez LA, Duchowny M. Carbamazepine toxicity resulting from generic substitution. Neurology 1993;43:2696–7.
4. Halkin H, Shapiro J, Kurnik D, Loebstein R, et al. Increased warfarin doses and decreased international normalized ratio response after nationwide generic switching. Clin Pharmacol Ther 2003;74:215–21.
5. Hartley R, Aleksandrowicz J, Bowmer CJ, Cawood A, et al. Dissolution and relative bioavailability of two carbamazepine preparations for children with epilepsy. J Pharm Pharmacol 1991;43:117–9.
6. Kluznik JC, Walbek NH, Farnsworth MG, Melstrom K. Clinical effects of a randomized switch of patients from clozaril to generic clozapine. J Clin Psychiatry 2001;62(Suppl 5):14–7.
7. Koch G, Allen JP. Untoward effects of generic carbamazepine therapy. Arch Neurol 1987;44:578–9.
8. Krämer G, Elger C, Denig D, Neubauer BA. Aut-idem-Ankreuzen: bei Antiepileptika wichtiger denn je! Aktuelle Neurologie 2008;335:108–9.
9. Krämer G, Schneble H, Wolf P. Risiken der neuen Aut-idem-Regelung für die Behandlung mit Antiepileptika. Aktuelle Neurologie 2002;29:115–22.
10. Mofsen R, Balter J. Case reports of the reemergence of psychotic symptoms after conversion from brand-name clozapine to a generic formulation. Clin Ther 2001;23:1720–31.
11. Novo-Venlafaxine XR, Product Monograph. Novo-Venlafaxine XR – Product Monograph. CR 9427-N0454/7-23. Document released under the Access to Information Act of Canada. 2006.
12. Nuss P, Taylor D, De HM, Hummer M. The generic alternative in schizophrenia: opportunity or threat? CNS Drugs 2004;18:769–75.
13. Pedersen SA, Dam M. [Carbamazepine: are synonymous preparations identical?]. Ugeskr Laeger 1985;147:2676–7.
14. Sachdeo RC, Belendiuk G. Generic versus branded carbamazepine. Lancet 1987;1:1432.
15. Simmenroth-Nayda A, Hummers-Pradier E, Ledig T, Jansen R, et al. Verordnung von Generika in der hausärztlichen Praxis. Ergebnisse einer Befragung von Hausärzten. Med Klin 2006;101:705–10.
16. Tschabitscher D, Platzer P, Baumgartel C, Mullner M. [Generic drugs: quality, efficacy, safety and interchangeability]. Wien Klin Wochenschr 2008;120:63–9.
17. Van AM, Mancini C, Patterson B, Bennett M. Symptom relapse following switch from Celexa to generic citalopram: an anxiety disorders case series. J Psychopharmacol 2007;21:472–6.
18. Welty TE, Pickering PR, Hale BC, Arazi R. Loss of seizure control associated with generic substitution of carbamazepine. Ann Pharmacother 1992;26:775–7.
Prof. Dr. med. Hans-Peter Volz, Krankenhaus für Psychiatrie, Psychotherapie und Psychosomatische Medizin Schloss Werneck, Balthasar-Neumann-Platz 1, 97740 Werneck, E-Mail: hans-peter-volz@kh-schloss-werneck.de
Prof. Dr. med. Jürgen Staedt, Klinik für Psychiatrie und Psychotherapie – Memory Clinic, Vivantes Klinikum Berlin-Spandau, E-Mail: Psychiatrie.Spandau@vivantes.de
Prof. Dr. med. Ion-George Anghelescu, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, Klinik und Hochschulambulanz für Psychiatrie und Psychotherapie, Eschenallee 3, 14050 Berlin (Ortsteil Charlottenburg), E-Mail: ion.anghelescu@charite.de
Prof. Dr. med. Eckart Rüther, Ludwig-Maximilians-Universität, Klinik für Psychiatrie und Psychotherapie, Nussbaumstraße 7, 80336 München, E-Mail: eruethe@gwdg.de
Psychopharmakotherapie 2009; 16(05)