Benedikt Schoser, München, Claudia Sommer, Würzburg, und Theo Dingermann, Frankfurt/M.
In den Lysosomen kernhaltiger Körperzellen befinden sich viele verschiedene Hydrolasen, deren Aufgabe es ist, komplexe Makromoleküle in ihre Bausteine zu zerlegen. Diese Enzyme sind auf bestimmte Substrate spezialisiert. Ist die Aktivität eines dieser Enzyme reduziert oder wird das Enzym gar nicht produziert, zum Beispiel aus genetischen Gründen, akkumuliert das jeweilige Substrat. Folge sind Störungen der Zellfunktion, später der Funktion der betroffenen Organe bis hin zum Organversagen. Je nachdem, welches dieser Enzyme bei den lysosomalen Speicherkrankheiten vermindert produziert wird, sind unterschiedliche Zelltypen/Organsysteme mehr oder weniger betroffen. Individuell bei jedem Patienten hängt die Ausprägung der Symptomatik bis zu einem gewissen Grad von den jeweiligen Mutationen der Gene ab, die das Enzym-Protein kodieren, sowie von der Restaktivität des Enzyms. Alle Faktoren für die phänotypische Ausprägung sind noch nicht hinreichend geklärt. Die Krankheitssymptome treten häufig bereits in der Kindheit und Jugend auf, werden jedoch oft falsch gedeutet oder aufgrund des zum Teil schleichenden Verlaufs zunächst wenig beachtet. Dennoch muss man konstatieren, dass die Diagnostik der lysosomalen Speicherkrankheiten insofern schwierig ist, als sie selten sind und sich mit sehr variablen Symptomen präsentieren. Sobald erst einmal die Idee differentialdiagnostisch aufkommt, dass es sich um eine lysosomale Speichererkrankung handeln könnte, ist die Diagnose mit La-bortests relativ rasch gesichert oder ggf. ausgeschlossen. Nicht selten haben die Patienten zum Zeitpunkt der Diagnose schon das Erwachsenenalter erreicht und es liegen bereits irreversible Organschäden vor. Vorrangiges Ziel muss es deshalb sein, das Intervall zwischen dem Auftreten erster Symptome und der Diagnosesicherung deutlich zu verkürzen. Dies und die frühzeitig eingesetzte Enzymersatztherapie, soweit für die einzelne lysosomale Speicherkrankheit vorhanden, könnte die Prognose vieler Patienten verbessern.
Morbus Pompe
Morbus Pompe (Glykogenose Typ II), benannt nach dem Erstbeschreiber, dem niederländischen Pathologen Joannes Cassianus Pompe (1901–1945), beruht auf einem Mangel des Enzyms saure Alpha-Glukosidase (GAA, auch: saure Maltase). In die Lysosomen gelangtes Glykogen wird normalerweise von der GAA zu Glucose hydrolysiert, die dann die Zellorganelle wieder verlassen kann. Der bei M. Pompe genetisch bedingte Mangel an lysosomaler GAA lässt den Glykogengehalt in den Lysosomen deutlich steigen. Andere zelluläre Strukturen werden dadurch verdrängt, später rupturieren die Lysosomen, wodurch weitere Enzyme ins Zytoplasma freigesetzt werden. Bei M. Pompe akkumuliert das Glykogen vor allem in der Skelett- Atem-und Herzmuskulatur, wodurch (unbehandelt) zunehmend Muskelzellen untergehen.
Häufigkeit und Genetik
In Europa erkrankt schätzungsweise einer von 40000 Menschen an M. Pompe. Je nach ethnischer Gruppe und Region werden Erkrankungshäufigkeiten zwischen 1/14000 und 1/300000 angegeben [5, 9]. Die Krankheit wird autosomal-rezessiv vererbt und basiert auf Mutationen im Gen für die GAA auf Chromosom 17. Es sind bei betroffenen Patienten bereits mehr als 250 Mutationen des Gens identifiziert worden. Die Überträger der Krankheit sind in der Regel heterozygot, selbst also klinisch gesund. Statistisch gesehen erhält ein Viertel ihrer Nachkommen beide mutierte Allele und erkrankt, ein weiteres Viertel erhält beide gesunden Allele und ist damit völlig gesund. Weitere 50% bekommen ein normales und ein mutiertes Allel vererbt, sind also potenzielle Überträger der Pompe-Krankheit, erkranken aber nicht [8–10].
Verlaufsformen und typische Symptome
Auch wenn infantile, juvenile und adulte Verläufe unterschieden werden, handelt es sich in der Realität doch um ein Kontinuum von Symptomen und Komplikationen. Es gilt die Faustregel: Je später die Krankheit beginnt, desto leichter verläuft sie. Kinder mit der klassischen infantilen Verlaufsform zeigen bereits in den ersten Lebenswochen eine ausgeprägte Muskelschwäche (floppy baby), Trinkschwäche, Schluck- und Gedeihstörungen. Der Mund steht zeltförmig offen, die Zunge ist groß, beim Anheben des Oberkörpers durch Zug an den Armen fällt der Kopf nach hinten und die Muskeleigenreflexe sind abgeschwächt. Diese Kinder versterben innerhalb des ersten Lebensjahrs, vor allem infolge der ausgeprägten Kardiomegalie mit Obstruktion der linksventrikulären Ausflussbahn oder an einer Aspirationspneumonie. Es gibt jedoch auch einen weniger foudroyanten infantilen Verlauf [8–10].
Bei der juvenilen Verlaufsform tritt die Symptomatik erst allmählich im Kleinkind- oder Schulalter zutage. Bei der adulten Verlaufsform bewirkt die noch vorhandende restliche Enyzmaktivität schleichend zunehmende Manifestationen jenseits des 30. Lebensjahrs. Das Herz ist bei juvenilen Pompe-Patienten selten, beim adulten Verlauf nicht oder allenfalls minimal betroffen.
Leitsymptome bei juvenilen/adulten Pompe-Patienten sind vor allem die Gliedergürtelschwäche, die Schwäche der Rumpfhaltemuskulatur sowie die neuromuskulär bedingte Atemschwäche. Die Kinder sind durchaus bewegungsfreudig, jedoch aufgrund der fehlenden Muskelkraft in ihren Leistungen eingeschränkt. Meist entwickeln sie eine Skoliose. Es treten Kontrakturen an Hüften, Knien und Sprunggelenken auf. Den Erwachsenen fällt zunächst körperliche Aktivität immer schwerer. Später wird das Aufstehen aus dem Sessel oder aus liegender Position zum Problem. Dies kann bei der klinischen Untersuchung zum Beispiel an einem positiven Gowers-Zeichen beim Aufrichten aus der Hocke oder beim Aufstehen aus dem Bett beobachtet werden (Abb. 1).
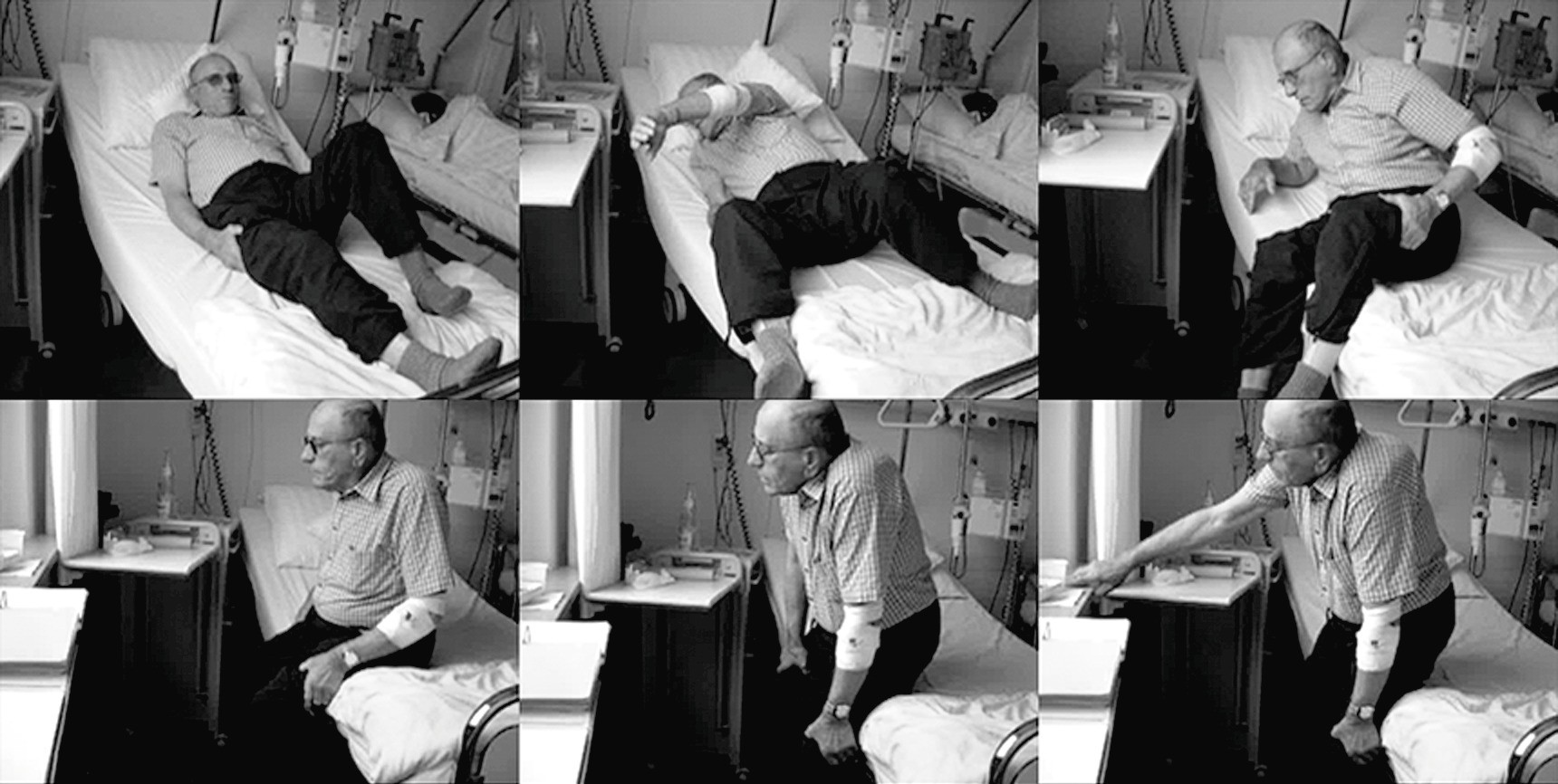
Abb. 1. Gowers-Manöver bei adultem M. Pompe; der Patient stützt sich an den eigenen Schenkeln ab, um sich aufrichten zu können [Foto: Schoser]
Erwachsene wie auch Kinder lassen einen Trendelenburg-Gang (Watschelgang) erkennen. Die sekundären, muskulär bedingten Atemprobleme führen nachts zu Hypoventilation oder Apnoe mit möglicher Sauerstoffentsättigung und Hyperkapnie des Blutes, Schlafstörungen, folgender Tagesmüdigkeit und Abgeschlagenheit. Die Atemmuskelschwäche lässt sich gut durch Messung der Vitalkapazität im Sitzen und im Liegen demonstrieren. Ein Abfall der forcierten Vitalkapazität um mehr als 20% ist dabei pathologisch. Zeitweise kann eine paradoxe Atembewegung im Liegen beobachtet werden. Die Ruheatemfrequenz ist erhöht, der Hustenstoß ist im Liegen deutlich schwächer ausgeprägt als im Sitzen [8, 10].
Ein Übersicht über klinische Charakteristika des M. Pompe gibt Tabelle 1.
Tab. 1. Klinische Charakteristika der Glykogenspeichererkrankung Typ 2 (M. Pompe) nach dem Alter der Erstmanifestation [modifiziert nach 8]
|
Infantile Form |
Juvenile Form |
Adulter Beginn |
|
|
Erkrankungsbeginn |
|||
|
<2 Jahre |
99% |
58% |
0% |
|
2.–15. Jahr |
1% |
42% |
0% |
|
>15 Jahre |
0% |
0% |
100% |
|
Muskelschwäche |
96% |
100% |
100% |
|
Kardiomegalie |
95% |
4% |
0% |
|
Hepatomegalie |
82% |
29% |
4% |
|
Makroglossie |
62% |
8% |
4% |
Morbus Fabry
Der Morbus Fabry, benannt nach dem deutschen Dermatologen Johannes Fabry (1860–1930), hat seine Ursache in der verminderten oder fehlenden Aktivität des Enzyms Alpha-Galactosidase A. Dieses lysosomale Enzym baut Glykosphingolipide ab – wichtige Membrankomponenten von Zellorganellen. Bei mangelnder Alpha-Galactosidase-A-Aktivität werden diese Glykosphingolipide, vor allem GL-3 (Globotriaosylceramid), zunehmend gespeichert, und zwar vor allem in den Gefäßendothelien, in allen Typen renaler Zellen, in Kardiomyozyten und im Reizleitungssystem sowie in Zellen des peripheren und zentralen Nervensystems. Folge sind der allmähliche Funktionsverlust der betroffenen Zellen und letztendlich schwere Endorganschäden an den Nieren, am Herzen und am Zentralnervensystem (Abb. 2). Dies schränkt die Lebenserwartung deutlich ein [2, 3, 12].
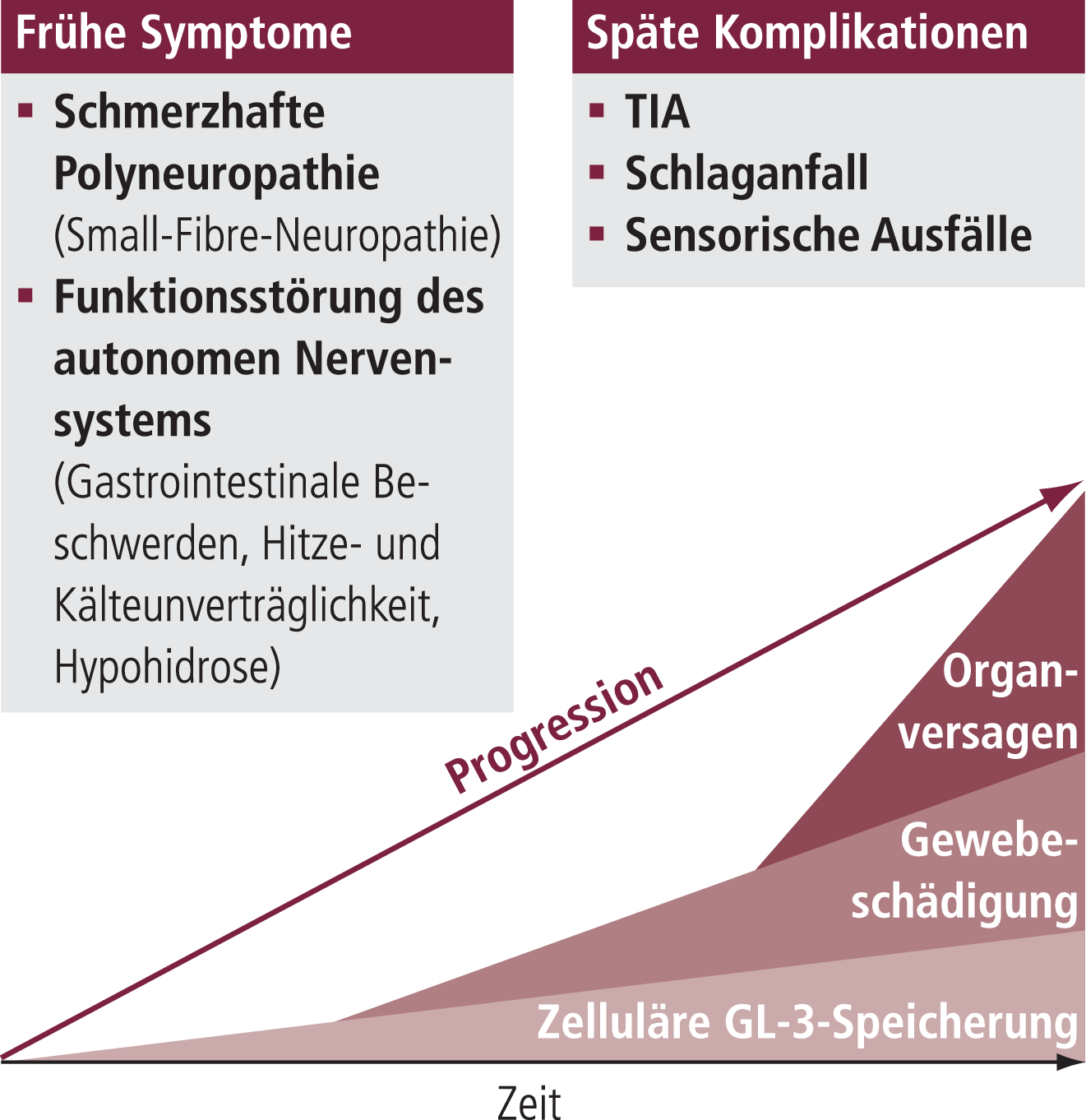
Abb. 2. Krankheitsmodell bei M. Fabry [modifiziert nach Wanner, C. Fabry Disease Model. Clinical Therapeutics; Vol. 29, Supplement A, 2007:2–5]
Häufigkeit und Genetik
Die Fabry-Krankheit wird X-chromosomal vererbt. Deshalb ging man lange davon aus, dass vor allem Männer betroffen sind. Bei männlichen Neugeborenen wird die Häufigkeit mit 1:40000 angegeben. Inzwischen weiß man, dass auch heterozygote Frauen betroffen sind und leichte bis schwere Symptome aufweisen können und damit auch eine verkürzte Lebenserwartung haben können. Offenbar wird eines der beiden X-Chromosomen bei diesen Frauen während der embryonalen Entwicklung unterschiedlich stark inaktiviert. Warum die Symptome bei Frauen so variabel sind, ist jedoch ungeklärt [6, 12].
Typische Symptome
Die geschilderte Pathogenese macht klar, dass die Krankheit viele Organsysteme einbezieht. Wie bei anderen lysosomalen Speicherkrankheiten auch, werden frühe Symptome, gerade im Kindes- und Jugendalter, häufig fehlgedeutet. Die Diagnosesicherung erfolgte bislang oft erst nach 14 bis 19 Jahren, so Daten aus dem Fabry-Register [3]. Aus neurologischer Sicht sollten unklare neuropathische Schmerzen nach Ausschluss von Diabetes mellitus, Alkoholabusus, Vitamin-B12-Mangel, entzündlicher und toxischer Ursachen den Diagnostiker aufmerken lassen. Hinzu kommen Kopfschmerzen, Konzentrationsstörungen sowie transitorische ischämische Attacken (TIA) und Schlaganfälle, oft bereits im frühen Erwachsenenalter. In einer Studie mit 33 Fabry-Patienten war festgestellt worden, dass bereits ein Viertel der Patienten im Alter von durchschnittlich 29 Jahren einen ersten Schlaganfall erlitten hatte [4].
Die Beschwerden, mit denen sich Fabry-Patienten typischerweise vorstellen, sind folgende:
- Schubweise auftretende brennende Schmerzen an Händen und Füßen, oft getriggert von körperlicher Aktivität, Hitze oder Kälte; auch Fieber tritt in solchen Phasen auf
- Kolikartige gastrointestinale Schmerzen, teilweise mit Übelkeit, Erbrechen und Durchfällen (infolge Störung des autonomen Nervensystems)
- Wärmeintoleranz und verminderte Schweißbildung
- Rötliche oder blau-schwärzliche, flache bis leicht erhabene Angiome, vor allem im Bereich des Bauchnabels, im Genitalbereich, an Mundschleimhaut und Konjunktiven (Abb. 3)
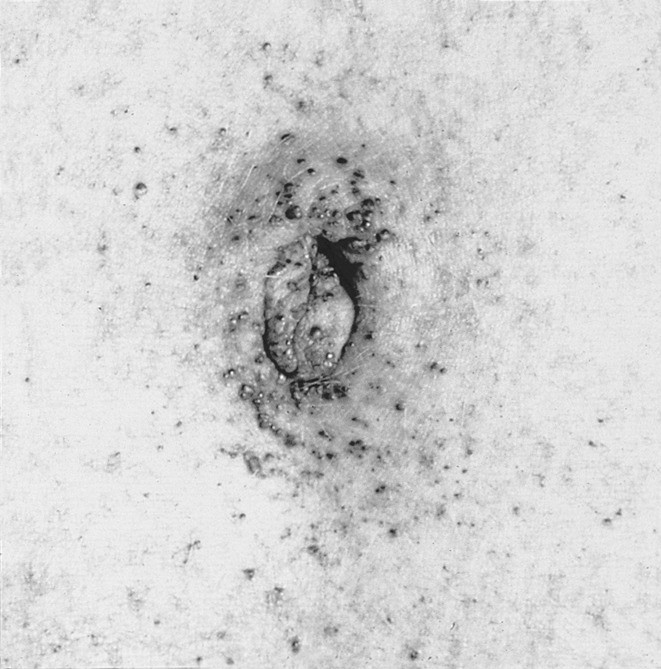
Abb. 3. Rötliche oder blau-schwärzliche, flache bis leicht erhabene Angiome [mit freundlicher Genehmigung von R.J. Desnick, PhD, MD, New York, USA]
Bei einer augenärztlichen Untersuchung können pathognomonische wirbelförmige Trübungen der Hornhaut (Cornea verticillata), stark geschlängelte Bindehautgefäße und weitere Veränderungen festgestellt werden (Abb. 4).
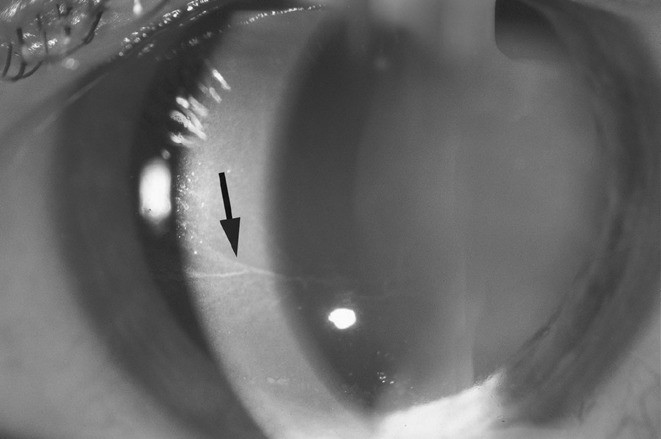
Abb. 4. Pathognomonische wirbelförmige Trübungen der Hornhaut, sog. Cornea verticillata [mit freundlicher Genehmigung von Prof. Dr. I. Lanzl, München]
Eine Polyneuropathie, die vor allem die kleinen Nervenfasern betrifft (Small-Fiber-Neuropathie der C- und A-delta-Fasern), manifestiert sich durch Störungen der Temperaturempfindung. Ein Zusammenhang zwischen den oben genannten Brennschmerzen und der Small-Fiber-Neuropathie wird vermutet. Im Verlauf kann eine zunehmende Dyspnoe der Patienten die progrediente Herzinsuffizienz anzeigen. Es treten pektanginöse Beschwerden, Palpitationen und bradykarde Herzrhythmusstörungen auf [11]. Häufig kommt es zu einer progredienten Niereninsuffizienz bis zur Dialysepflichtigkeit; erste Anzeichen sind Proteinurie und verminderte glomeruläre Filtrationsrate (GFR).
Diagnosesicherung
Die Diagnose von M. Pompe und M. Fabry oder anderen lysosomalen Speicherkrankheiten ist im Grunde einfach, sobald man an diese mögliche Diagnose gedacht hat. Alle diese Krankheiten beruhen auf messbar verminderten Enzymaktivitäten. Im Allgemeinen gehen dem andere Untersuchungen und Maßnahmen zur Eingrenzung der infrage kommenden Diagnose voran.
So erkennt man in der Elektromyographie (EMG) von Pompe-Patienten myogene Veränderungen motorischer Einheiten und gelegentlich langanhaltende myotone Serienentladungen. Die Magnetresonanztomographie zeigt einen fettigen Umbau der Muskulatur, teilweise mit erheblichen Flüssigkeitseinlagerungen zum Beispiel in der sogenannten ischiocruralen Muskulatur der Oberschenkel (Kennmuskel M. biceps femoris). Im Muskelbiopsat findet der Pathologe große Vakuolen in den Muskelzellen mit glykogenhaltigem Inhalt in der PAS-Färbung. Erhöhte Leberwerte lassen bei manchen Patienten zunächst an eine primäre Lebererkrankung denken, wenn nicht parallel bereits die Creatinkinase (CK) mitbestimmt worden ist. Die CK ist beim klassischen infantilen M. Pompe massiv erhöht, aber auch fast immer bei juvenilen/adulten Pompe-Patienten. Herzuntersuchungen, angefangen beim Röntgen-Thorax, dem Elektrokardiogramm bis hin zur Herzechokardiographie, lassen die teilweise erhebliche Kardiomegalie bei infantilen Verlaufsformen oder die hypertrophe Kardiomyopathie erkennen. Häufige Fehldiagnosen bei Pompe-Patienten sind unklare Myopathien, Krankheiten des rheumatischen Formenkreises und funktionelle Störungen.
Auch die oben beschriebene variable Symptomatik bei Fabry-Patienten lässt zunächst an andere Ursachen denken, so bei neuropathischen Schmerzen an Diabetes mellitus, Alkoholabusus, Vitamin B12-Mangel, entzündliche oder toxische Ursachen, an kardiovaskuläre Krankheiten anderer Ursache oder bei starker gastrointestinaler Symptomatik an Appendizitis, Steinleiden oder andere urologische und gastrointestinale Krankheitsbilder. Deshalb gilt es, bei unklaren Verläufen und Symptommustern oder bei unzureichenden Therapieerfolgen immer wieder die Arbeitsdiagnose zu hinterfragen.
Als diagnostischer Standard bei M. Pompe gilt derzeit die Bestimmung der GAA-Restaktivität aus Lymphozyten aus einer EDTA-Blutprobe. Bei schweren infantilen Formen eignet sich auch Trockenblut. Die GAA-Aktivitätsmessung kann darüber hinaus in pathologisch veränderter Muskulatur oder in kultivierten Hautfibroblasten erfolgen [5, 7, 8].
Bei Verdacht auf M. Fabry wird die Diagnose gesichert, indem die Alpha-Galactosidase-A-Aktivität im Plasma oder in peripheren Lymphozyten bestimmt wird. Bei Frauen muss eventuell zusätzlich die molekulargenetische Untersuchung vorgenommen werden, weil bei ihnen unter Umständen eine normale Enzymaktivität gemessen wird, obwohl eindeutige Fabry-Symptome bestehen. Zur Gesamtdiagnostik dieser lysosomalen Speicherkrankheiten sowie zur folgenden Therapie und zum Monitoring liegen detaillierte Expertenempfehlungen vor [2, 3, 5, 11].
Die Behandlung mit Enzymersatztherapien
Seit 2001 ist die kausale Enzymersatztherapie für Fabry-Patienten verfügbar, seit 2006 gibt es die kausale Enzymersatztherapie für Pompe-Patienten. Es handelt sich dabei um jeweils in aufwendigen Verfahren gentechnisch hergestellte Produkte, die im Allgemeinen alle zwei Wochen infundiert werden. Für M. Fabry sind in Europa mit Agalsidase alfa (Replagal®) und Agalsidase beta (Fabrazyme®) zwei Arzneimittel auf dem Markt.
Ziel ist es, die gespeicherten Glykosphingolipide allmählich abzubauen und damit die klinischen Symptome zu lindern sowie die organspezifischen Komplikationen zu verhindern. In einer internationalen Multicenterstudie mit 82 Patienten ist zum Beispiel für Agalsidase beta nachgewiesen worden, dass der Krankheitsprogress über 35 Monate im Vergleich zur Plazebo-Behandlung aufgehalten werden kann. Das Risiko für das Auftreten renaler, kardialer und zerebrovaskulärer Komplikationen konnte um mehr als die Hälfte verringert werden [1].
Für Pompe-Patienten ist die rekombinante Alglucosidase alfa (Myozyme®) verfügbar. In klinischen Studien konnte bei Säuglingen im Alter von weniger als sechs Monaten die Lebensdauer und das beatmungsfreie Überleben verlängert werden, die Kardiomyopathie, die motorische Leistung und das Wachstum verbesserten sich [7]. Individuell ist im Moment der langfristige Verlauf jedoch schwer vorherzusagen [7]. Bei juvenilen und adulten Pompe-Patienten lässt sich im Vergleich zu Plazebo-behandelten Patienten eine Stabilisierung der Atemfunktion und der Gehfähigkeit verzeichnen [7,10]. Mit einem frühzeitigen Therapiebeginn scheinen die besten Resultate zu erreichen zu sein, denn einmal eingetretene Endorganschäden sind irreversibel.
Wichtig ist nach wie vor die Begleittherapie in Form von Physiotherapie und Muskeltraining, atemunterstützenden Maßnahmen, proteinreicher Ernährung und Schmerztherapie.
Biotechnologie
Bei den Wirkstoffen, die in der Enzymersatztherapie eingesetzt werden, handelt es sich um sehr große und komplexe Proteine, die nicht wie herkömmliche Arzneimittel chemisch synthetisiert werden können. Vielmehr lässt man diese Proteine von Mikroorganismen oder geeigneten Zelllinien „nachbauen“. Dies ist möglich, weil der genetische Code universell ist: Der genetische Bauplan eines humanen Enzyms kann in die Erbsubstanz nichthumaner Zellen integriert und dort ebenso gelesen und verstanden werden. Diese gentechnisch produzierten Proteine sind sehr viel sicherer als analoge Proteine, die man früher direkt aus dem Menschen isoliert hat. Denn Letzteres birgt gewisse Restrisiken für die Übertragung von Infektionserregern. Das ist bei gentechnisch hergestellten Medikamenten so gut wie ausgeschlossen.
Eine Herausforderung bei einer Enzymersatztherapie für M. Fabry und M. Pompe ist die Tatsache, dass die hergestellten Proteine nicht nur einfach ins Blut des Patienten gelangen müssen, sondern über spezielle Rezeptoren vermittelt in die betroffenen Zellen hinein. Dies gelingt ihnen mit Hilfe von Zuckerstrukturen auf der Enzymoberfläche, die mit Rezeptoren an den Zielzellen interagieren, so dass die Enzyme über diese Rezeptoren in die Zellen aufgenommen werden. Die Informationen für diese Zuckerstrukturen sind jedoch nicht in der DNS abgelegt, sondern sind organismusspezifisch. Deshalb lassen sich diese Wirkstoffe nur in ganz bestimmten Zellen herstellen, die die Zucker korrekt an das Enzym-Protein ansynthetisieren können. Die unnatürliche Enzymzufuhr von extern macht demnach Veränderungen an den Makromolekülen erforderlich, so dass viele biotechnisch hergestellte Medikamente nicht den körpereigenen „Originalen“ entsprechen können.
All dies macht den Herstellungsprozess von Enzymersatztherapeutika äußerst aufwendig. Zunächst muss der genetische Bauplan für das Enzym in die Wirtszelle, etwa Ovarialzellen des chinesischen Hamsters (CHO-Zellen), integriert und ein entsprechender Zellklon selektioniert werden. Dieser Zellklon ist der Ausgangspunkt für die Herstellung einer Masterzellbank, für die die Arzneimittelbehörden eine Zulassung erteilen. Daraus werden wiederum Arbeitszellbanken generiert und diese Zellen schrittweise in immer größeren Fermentern kultiviert (100 bis 2000 bzw. 4000 Liter). Die Zellen sezernieren das therapeutische Protein in das Kulturmedium, so dass man die Zellen nicht aufbrechen muss, um an das Enzym zu gelangen. Es folgen mehrere Aufreinigungsschritte, alles unter strenger Überwachung und der Kontrolle hunderter Parameter. Anschließend muss das gewonnene Makromolekül noch durch biochemische Prozesse bearbeitet werden, damit es, ohne zerstört zu werden, an seinen Wirkort gelangen und dort auch tatsächlich seine Wirkung entfalten kann. Erst nach einem weiteren Aufreinigungsverfahren kann der komplexe Wirkstoff zu einer stabilen Formulierung verarbeitet und portioniert werden (Abb. 5).
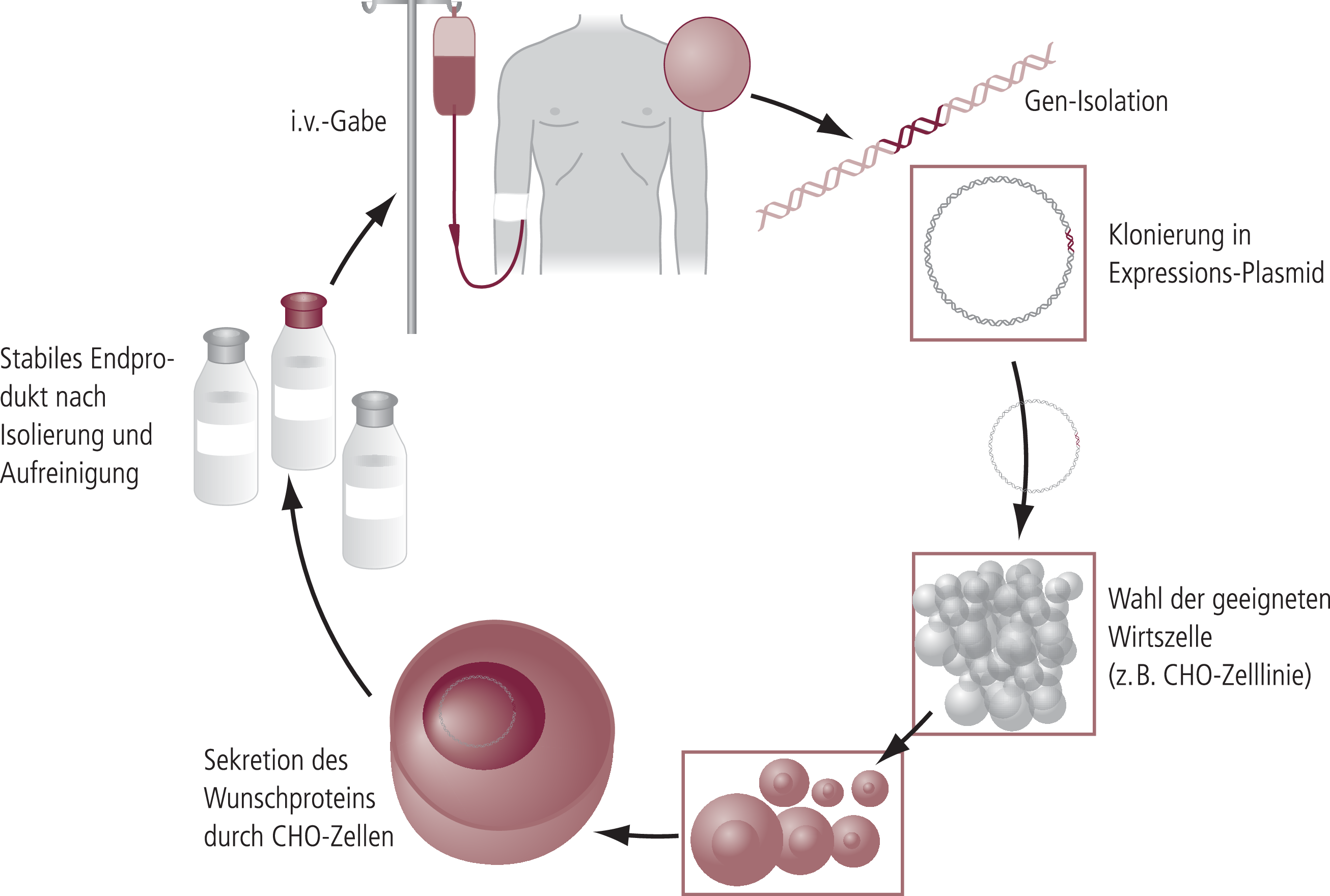
Abb. 5. Gentechnische Herstellung therapeutischer Proteine (z.B. Enzyme) – Übersicht
Anders als bei chemisch synthetisierten Medikamenten, deren Zusammensetzung normalerweise stets einheitlich und über chemische und physikalische Parameter definiert ist, werden gentechnisch hergestellte Wirkstoffe zusätzlich über die Herstellungsprozesse charakterisiert. Diese Prozesse sind bis ins kleinste Detail definiert, um trotz der Isolation der Moleküle aus einer hochkomplexen Matrix Arzneimittelchargen mit weitgehend identischer Wirksamkeit zu generieren. Der Prozess bestimmt also das Produkt, der Prozess ist das Produkt.
Fazit
Einige der seltenen lysosomalen Speicherkrankheiten wie Morbus Pompe und Morbus Fabry gehen mit neurologischen Symptomen einher, die bei genauer Betrachtung zur Diagnose führen können. Die Erkrankungen beruhen auf angeborenen Enzymdefekten, die mit einfachen Labortests nachgewiesen werden können. Für die genannten Stoffwechselerkrankungen gibt es bereits Enzymersatztherapien, die im Allgemeinen umso effektiver sind, je früher sie eingesetzt werden und bevor irreversible Schäden eingetreten sind. Diese Enzymersatzpräparate werden biotechnisch in sehr aufwendigen, streng kontrollierten Verfahren hergestellt.
Literatur
1. Banikazemi M, et al. Agalsidase-beta therapy for advanced Fabry disease. Ann Intern Med 2007;146:77–86.
2. Eng CM, et al. Fabry disease: Guidelines for the evaluation and management of multi-organ system involvement. Genet Med 2006;8: 539–48.
3. Eng CM, et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the Fabry-Registry. J Inherit Metab Dis 2007;30:184–92.
4. Grewal RP. Stroke in Fabry’s disease. J Neurol 1994;241:153–6.
5. Kishnani PS, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8:267–88.
6. MacDermot KD, et al. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001;38:769–75.
7. Schoser B, Hill V, Raben N. Therapeutic approaches in glycogen storage disease type II/Pompe Disease. Neurotherapeutics 2008;5: 569–78.
8. Schrank B. CME Morbus Pompe/Glykogenose Typ II. Stuttgart: Thieme, 2009.
9. Schwarz M, Berger M. Kohlenhydratstoffwechsel und Glykogenosen in: Strohmeyer G, Stremmel W, Niederau C (Hrsg.): Angeborene Stoffwechselerkrankungen. Landsberg: ecomed, 2002:29–31.
10. van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet 2008;372:1342–53.
11. Weidemann F, Breunig F. Kardiale Beteiligung beim Morbus Fabry. Med Klinik 2008;103:161–5.
12. Wilcox WR, et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol Genet Metab 2008;93:112–28.
Priv.-Doz. Dr. med. Benedikt Schoser, Friedrich-Bauer-Institut, Neurologische Klinik, Ludwig-Maximilians-Universität München, Ziemssenstr. 1a, 80336 München, E-Mai:l bschoser@med.uni-muenchen.de
Prof. Dr. med. Claudia Sommer, Neurologische Klinik und Poliklinik des Universitätskllinikums Würzburg, Josef-Schneider-Straße 11, 97080 Würzburg
Prof. Dr. rer. nat. Theo Dingermann, Institut für Pharmazeutische Biologie, Theodor-Stern-Kai 7, Johann Wolfgang Goethe-Universität Frankfut, 60590 Frankfurt a. M.
Morbus Pompe and Morbus Fabry: rare treatable metabolic diseases
Within the group of rare lysosomal storage disorders, there are diseases with manifestations of primary neurological deficits or syndromes. This report will focus on clinical aspects, diagnostic work-up and therapy of two hereditary lysosomal storage diseases, Pompe and Fabry disease. Often both diseases need an interdisciplinary approach for making the correct diagnosis. Pompe disease clinically manifests primary as a skeletal muscle disorder with muscle weakness, respiratory insufficiency and cardiomyopathy. In infantile Pompe disease, babies already present as floppy infants at birth. In juvenile and adult patients, motor mile stones are prolonged, and later patients will be become dependent on walkers and wheelchairs and/or noninvasive respiratory devices. Fabry patients frequently present with attacks of burning painful sensations at hands and feet. Commonly, the cause of these symptoms resides uncovered over many years. Furthermore, the autonomic nervous system is regularly altered in Fabry patients.
However, both diseases have become prototypes for advances in modern biotechnology, which tries to turn untreatable hereditary disorders into treatable entities. This opens prognostic hopeful avenues for patients with rare diseases. During a complex manufacturing using gene and cell line culture techniques, macromolecules have be produced in order to substitute the missing enzyme in Pompe or Fabry patients. Today, this new enzyme replacement therapy can help to stabilize and partly improve clinical symptoms in many Pompe and Fabry patients.
Key words: Pompe disease, Fabry disease, biotechnology, enzyme replacement therapy
Psychopharmakotherapie 2009; 16(05)